Cascade of events leading to prion disease described
Prion diseases are deadly neurodegenerative disorders in humans and animals that are characterized by misfolded forms of prion protein (PrP). Development of effective treatments has been hampered by the lack of good experimental models. In a new study published in The American Journal of Pathology, researchers describe the distinct stages of prion disease in the mouse retina and define an experimental model to specifically test therapeutic approaches.
"This work was done in collaboration with the USDA Agricultural Research Service and is an excellent example of how animal disease research can be leveraged to benefit human health," commented Heather West Greenlee, PhD, an Associate Professor in the Department of Biomedical Sciences at the Iowa State University College of Veterinary Medicine (Ames IA). "It provides important insights into the timeline for key pathologic milestones of prion disease in the retina and a model to study mechanisms of disease progression and evaluate therapeutic interventions."
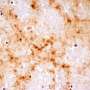
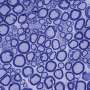
The study used an experimental mouse model of the prion disease scrapie to determine the temporal relationship between the transport of misfolded prion protein (PrPSc) from the brain to the retina, the accumulation of PrPSc in the retina, the inflammatory response of the surrounding retinal tissue, and the loss of neurons.
It is believed that transmissible spongiform encephalopathy (TSE) progression depends on the spread of misfolded protein from one central nervous system structure to another. The investigators injected mouse-adapted scrapie into the brains of mice and studied the movement of misfolded prion protein from the brain into the retina via the optic nerve for up to 153 days post-inoculation (dpi), when clinical disease becomes apparent.
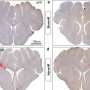
By studying prion disease in the retina, which is relatively isolated from the brain, researchers were able to determine the time lag between stages of the disease process and were able to sequentially detect seeding of misfolded protein in the retina at 60 dpi, followed by accumulation of PrPSc and activation of retinal glia at 90 days, activation of microglia at 105 dpi, and retinal neuronal death at 120 dpi.
Prion diseases, also known as transmissible spongiform encephalopathies (TSEs), are a transmissible protein misfolding disease, which makes them a good model to study common aspects of these diseases. "Not only will this work contribute to the development of therapy to treat prion disease, but it may also provide important insights into which therapies may be effectively applied to the treatment of other protein misfolding diseases, including Parkinson disease and Alzheimer disease," explained Dr. Greenlee.
Using this information, it is now possible to evaluate a potential therapy for its ability to interfere with accumulation of protein misfolding, to suppress damaging neuroinflammation, or to prevent death of neurons.
Prion diseases include Creutzfeldt-Jakob disease and kuru in humans, scrapie in sheep, mad cow disease in cows, and chronic wasting disease in deer.
More information: Temporal Resolution of Misfolded Prion Protein Transport, Accumulation, Glial Activation, and Neuronal Death in the Retinas of Mice Inoculated with Scrapie, The American Journal of Pathology, DOI: 10.1016/j.ajpath.2016.05.018