Common molecular mechanism of Parkinson's pathology discovered
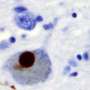
 of an intraneural Lewy-body in the Substantia nigra in Parkinson's disease. Credit: Wikipedia")
Stanford University School of Medicine researchers have located an intracellular defect that they believe is probably common to all forms of Parkinson's disease.
This defect, which precedes the death of a group of nerve cells whose loss is the hallmark of the condition, play a critical role in triggering that die-off.
"We've found a molecular biomarker that characterizes not just familial cases of Parkinson's, in which a predisposition for the disease is clearly inherited, but also the condition's far more prevalent sporadic forms, for which the genetic contribution is either nonexistent or not yet discovered," said Xinnan Wang, MD, PhD, assistant professor of neurosurgery.
The defect, described in a study to be published online Sept. 8 in Cell Stem Cell, renders cells unable to quickly dismantle their internal power packs, called mitochondria, when they wear out, stop supplying energy and start spewing out pollutants instead. This discovery could lead to not only more accurate but much earlier diagnoses of Parkinson's disease and could also point to entirely new pharmacological approaches to treating it, said Wang, who is the study's senior author. The lead author is postdoctoral scholar Chung-Han Hsieh, PhD.
The second-leading neurodegenerative disorder after Alzheimer's disease, Parkinson's affects one in every 60-70 Americans age 65 or older. While 5-10 percent of all cases are familial, the vast majority are sporadic.
Prevalent mutation in Parkinson's
The most frequent genetic mutations responsible for familial Parkinson's occur at various points along a gene coding for a protein called LRRK2. Several such mutations are known, but genetic tests reveal that the mutation known as LRRK2G2019S is the most prevalent, turning up in 1 in 20 of familial cases and 1 in every 50 apparently sporadic cases among Caucasians. Curiously, LRRK2G2019S shows up in 40 percent of familial Parkinson's cases and 13 percent of sporadic cases among Ashkenazi Jews; figures for North African Berbers are 39 and 40 percent, respectively.
Until now, no one could clearly account for LRRK2's connection to Parkinson's. In an extensive series of experiments with cells cultured from Parkinson's patients and healthy subjects, Wang and her colleagues have cleared up that mystery.
Mitochondria convert fats and sugars into other molecules that transport energy where it's needed in a cell. A single nerve cell can host hundreds or even thousands of mitochondria. A vast supply of energy is particularly crucial for a group of midbrain nerve cells whose nonstop activity fine-tunes our voluntary movements: These cells constantly produce and secrete a substance called dopamine (they're called "dopaminergic"), and each sends out numerous long-distance, dopamine-squirting cellular extensions to elsewhere in the brain. The failure of these dopamine-producing midbrain nerve cells triggers the classic symptoms of Parkinson's disease: tremor, stiffness, difficulty initiating and sustaining voluntary motion, and, sometimes, cognitive difficulties.
Until now, the question of what causes the death of these dopaminergic nerve cells in Parkinson's patients has occasioned many highly uncertain guesses backed by little solid evidence. This uncertainty limits medical practitioners' ability to diagnose Parkinson's early on, and it impedes drug developers' attempts to find effective treatments.
Malfunctioning mitochondria are like old jalopies: Their fuel efficiency is rotten, and they spew out tons of toxic exhaust in the form of corrosive chemicals called free radicals. But, the Stanford scientists showed, before faulty mitochondria can be decommissioned, they must first be detached from the cytoskeleton, a network of molecular filaments and tubules that spans and shapes most of our cells. Only after the mitochondria are detached can the cell destroy them. But this can't happen, Wang's team found, until a protein called Miro that anchors mitochondria to the cytoskeleton is severed.
Removing Miro
The researchers discovered that Miro's removal can occur only after LRRK2 forms a complex with Miro. Defective LRRK2 is impaired in forming this complex, resulting in significant delays in Miro's removal.
In the study, Wang and her associates obtained 20 different lines of cultured fibroblasts from human skin: four from healthy subjects; five from apparently sporadic Parkinson's patients; six from familial patients with LRRK2 mutations, including the notorious LRRK2G2019S; and five from familial patients with other mutations.
Six hours after biochemically inducing mitochondrial damage in these cells, the scientists broke open some of them to observe signs of Miro degradation. At 14 hours post-biochemical assault, they broke more cells open to monitor mitochondrial breakdown. In all four cultured fibroblast lines from healthy subjects, that's just what they witnessed. But to their surprise, mitochondrial detachment and breakdown were substantially delayed in all 16 cell lines representing Parkinson's cases.
Digging deeper, the scientists used advanced methods to create nerve cells enriched for dopaminergic activity from several of the skin-fibroblast lines.
They then employed techniques ranging from live imaging with microscopic cameras to biochemical manipulations to show that damaged mitochondria in the dopaminergic nerve cells generated from healthy subjects' fibroblasts quickly led to those mitochondria being destroyed. But in the dopaminergic nerve cells derived from the cells of patients with LRRK2G2019S-mutant Parkinson's, this process and the key steps leading up to it were substantially delayed.
Free radicals
When the researchers biochemically induced excessive free-radical production in the nerve cells, those from every Parkinson's patient sampled—familial and sporadic alike—died in much greater numbers than equivalent cells derived from healthy patients.
"The healthy cells could handle higher free-radical concentrations," Wang said. "But the Parkinson's cells were more prone to dying under those conditions, which are apt to occur in the energy-intensive, midbrain, dopamine-producing nerve cells that degenerate in Parkinson's disease."
Remarkably, the scientists discovered they could prevent the delay in Parkinson's-derived nerve cells' dismantling of faulty mitochondria, as well as forestall those cells' untimely death in the face of free-radical onslaught. They performed a biochemical trick that reduced Miro levels in the cells. The reduction wasn't enough to dislodge healthy mitochondria from the cytoskeleton, but it reduced their attachment intensities closer to the point at which detachment could occur. When the scientists then chemically induced mitochondrial damage, no increased mitochondrial drop off or degradation took place in the nerve cells derived from healthy subjects. But in the equivalent LRRK2G2019S nerve cells, the previously seen delays pretty much disappeared—and far fewer of these cells died. Lowering Miro concentrations, in those cells, compensated for their Miro-chopping impairment.
In a fruit-fly model of LRRK2 linked-Parkinson's locomotion difficulties (good rodent models for this aspect of Parkinson's don't exist), lowering Miro levels resulted in the restoration of larvae's visibly compromised crawling ability and fully reversed deficits in the climbing and jumping abilities of adult flies.
Wang's team also proved that LRRK2 is recruited to sites of mitochondrial stress. They believe many different intracellular difficulties can contribute to the failure of even a perfectly normal LRRK2 complex with Miro, and that Miro's resultant failure to release its grip may be a reliable early biomarker—and possibly the crucial causal event—of Parkinson's disease.
"Existing drugs for Parkinson's largely work by supplying precursors that faltering dopaminergic nerve cells can easily convert to dopamine," Wang said. "But that doesn't prevent those cells from dying, and once they've died you can't bring them back. Measuring Miro levels in skin fibroblasts from people at risk of Parkinson's might someday prove beneficial in getting an accurate, early diagnosis. And medicines that lower Miro levels could prove beneficial in treating the disease."
The work is an example of Stanford Medicine's focus on precision health, the goal of which is to anticipate and prevent disease in the healthy and precisely diagnose and treat disease in the ill.