Research suggests that chronic traumatic encephalopathy is a prion disease
A shared biological mechanism may drive the progression of both Alzheimer's disease (AD) and chronic traumatic encephalopathy (CTE), a neurodegenerative condition associated with repeated concussions and brain trauma, according to a new study led by UC San Francisco scientists.
Both AD and CTE are classified as "tauopathies," a category of diseases characterized by the improper folding and clumping together of a protein called tau (rhymes with "how") inside the nerve cells of the brain. The resulting tau aggregates, known as neurofibrillary tangles, are toxic to neurons and are thought to be responsible for the behavioral changes and cognitive decline seen in both disorders.
The senior author of the new study, Stanley Prusiner, MD, director of the Institute for Neurodegenerative Diseases (IND), part of the UCSF Weill Institute for Neurosciences, has long held that misfolded tau spreads through the brain because it forms prions, self-propagating proteins similar to those that cause diseases such as bovine spongiform encephalopathy (BSE, or "mad cow disease"). Prusiner was awarded the Nobel Prize in 1997 for discovering the role of prions in BSE and related diseases.
The new research, the first to document tau prions in CTE patients, made use of an experimental platform designed to test prion transmission in human cell cultures. As reported on November 28, 2016 in the online Early Edition of Proceedings of the National Academy of Sciences, misfolded tau from the brains of either AD or CTE patients propagated in these cell cultures and formed aggregates under identical conditions. But successful propagation of tau samples from patients with other neurodegenerative diseases, such as Pick's disease, a rare form of dementia that affects the brain's frontotemporal lobes, required different conditions.
"This work tells us that there are inherent differences, and sometimes similarities, among the tauopathies," said first author Amanda Woerman, PhD, assistant adjunct professor of neurology and a member of the IND. "As we develop new therapies to halt progression and neurodegeneration in these conditions, we may find that we need a drug specifically designed for both Alzheimer's and CTE, another for Pick's disease, and so on."
The CTE patient samples were provided by Ann McKee, MD, professor of neurology and pathology at Boston University School of Medicine, and a leader in the study of CTE in athletes and military veterans. Patient samples representing other tauopathies were made available by Lea T. Grinberg, MD, PhD, and William W. Seeley, MD, both associate professors of neurology and members of the UCSF Memory and Aging Center.
Tau normally stabilizes microtubules, long cylindrical structures that form the cell's internal scaffolding and help to transport various proteins. A section of the tau protein known as the repeat domain, so called because specific sequences of amino acids are repeated in this region, helps tau fulfill this stabilizing role by binding tightly to microtubules. The tau protein contains either 3 repeats (3R) or 4 repeats (4R) in this region.
Pick's disease is characterized by aggregates of 3R tau, while another neurodegenerative condition, called progressive supranuclear palsy (PSP), is associated with 4R aggregates. The aggregates seen in AD and CTE are composed of both 3R and 4R tau.
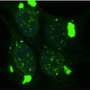
The cell culture platform relies on a human-derived cell line (HEK cells) carrying several copies of 3R tau, 4R tau, or both, each fused to a "reporter" molecule known as yellow fluorescent protein, or YFP. This approach was based on a cell line initially devised by former UCSF faculty member Marc Diamond, MD, now director of the Center for Alzheimer's and Neurodegenerative Diseases at UT Southwestern Medical Center in Dallas, Texas, which employed 4R tau exclusively.
These fused tau proteins serve as a "template" to test prion propagation: tau prions obtained postmortem from the brains of patients are added to the medium containing the engineered HEK cells, and if they cause the fused tau-YFP proteins to aggregate, the YFP emits a strong fluorescent signal that can be precisely measured. A great strength of this platform is that propagation can be reliably detected in as little as four days, an important methodological advantage in research on neurodegenerative diseases, which typically develop extremely slowly.
In the new study, tau prions from Pick's disease patients successfully propagated in HEK cells carrying 3R tau while tau prions from PSP patients successfully infected HEK cells expressing 4R tau. However, tau prions from AD or CTE patients did not propagate in either condition. Instead, propagation was successful only when the HEK cells expressed both 3R and 4R tau.
"We've known that tangles in the brains of both Alzheimer's and CTE patients are composed of both 3R and 4R tau," Woerman said. "What wasn't known before this study is whether these 3R and 4R tau proteins could propagate separately, or whether propagation requires the presence of both forms. Our work shows that the latter seems to be the case."
Woerman said these findings have implications for the development of new drugs, allowing scientists to test potential therapies against disease-specific tau prions to potentially determine which patients will respond to a given drug, and also for the creation of better diagnostic tools.
In just the past two years, for example, there has been great excitement surrounding the emergence of imaging probes that allow tau deposits in the brain to be detected by positron emission tomography (PET)—before this development, tau could only be measured in postmortem brain tissue. But Woerman said these probes work best in AD and not as well in other neurodegenerative diseases, and she suspects a better understanding of disease-specific tau isoforms, such as that documented in the new study, may be the key to creating more precise probes.
More information: Amanda L. Woerman et al. Tau prions from Alzheimer's disease and chronic traumatic encephalopathy patients propagate in cultured cells, Proceedings of the National Academy of Sciences (2016). DOI: 10.1073/pnas.1616344113