New compound targets TB bacterium's defense against the immune system

Part of the reason tuberculosis-causing bacteria are so good at colonizing the human body is that they have defenses against the body's immune system. A research team led by a Brown University chemist has developed a new compound that can take down one of those defenses in Mycobacterium tuberculosis. The researchers are hopeful that the compound could be part of a new drug strategy for treating tuberculosis.
"Given the increasing resistance of Mycobacterium tuberculosis to drugs, we contemplated the treatment of tuberculosis in a fundamentally different way," said Jason Sello, associate professor of chemistry at Brown who directed the research. "Instead of seeking conventional drug leads that kill M. tuberculosis directly, we hoped to develop compounds that could render the bacterium susceptible to the immune system. We were successful in designing compounds that make laboratory-grown bacteria sensitive to a chemical produced during the immune response."
Kyle Totaro, who recently earned his Ph.D. from Brown, led Sello's team. They also worked collaboratively with research groups at the Massachusetts Institute of Technology and Weill Cornell Medicine. A paper describing the work is published in the journal ACS Infectious Diseases.
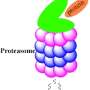
The team's strategy was to inhibit an enzyme found in M. tuberculosis called the 20S proteasome. It acts like a molecular trash collector, disposing of damaged proteins within the bacterial cell. It specializes in cleaning up proteins damaged by nitric oxide, a chemical produced by the innate immune system to help fight pathogens. The ability of the 20S proteasome to dispose of nitric oxide-damaged proteins helps the bacteria survive within the host.
To inhibit the proteasome, Sello and his team envisioned reactive compounds that mimic key chemical attributes of its substrates—the proteins that the enzyme normally breaks down. They anticipated that the proteasome would bind these compounds as it does any other protein and that their reactivity would disable the enzyme. With the proteasome disabled, proteins damaged by nitric oxide will accumulate inside the bacteria and cause their death.
But there was a key problem the researchers needed to contend with. Humans have a very similar system for the degradation of damaged proteins, and inhibition of this system is known to be lethal to cells. So any compound Sello and his team developed would have to selectively disrupt that bacterial proteasome, without significantly affecting the human version.
To do that, the researchers drew inspiration from nature. A bacterium called Pseudomonas syringae, a pathogen that infects plants, is known to produce compounds called syringolins, which are known to inhibit the plant proteasome by mimicking its substrates. The compounds are also known to inhibit the human proteasome and have promise as anticancer agents. Sello and his team used predictions about how the syringolins bind the human proteasome and knowledge about the substrates of the M. tuberculosis proteasome to design selective inhibitors.
Research had indicated that syringolins bound and inhibited the human proteasome by mimicking a preferred substrate have a specific chemical residue (valine) at two key positions. Research had also indicated that the bacterial proteasome prefers to degrade proteins having two different chemical residues (tryptophan and glycine) at the same two key positions. So, the researchers predicted that a syringolin analog in which the valine residue was swapped for structures resembling the tryptophan and glycine, the compound would selectively inhibit the bacterial proteasome.
Sello and his students at Brown synthesized the designed compound as well as others that matched or conflicted with their design model. In turn, their collaborators at MIT systematically assessed the capacities of the compounds to inhibit both the human and bacterial proteasomes in a test tube.
The team found that the natural syringolin product was 160-fold more specific for the human proteasome. One of the engineered syringolin analogs, in contrast, was 74-fold more specific for the bacterial proteasome.
"Using this rational design approach and chemical synthesis, we were able to generate selective inhibitors of the M. tuberculosis 20S proteaseome," Sello said. "In the best case, our engineering of the syringolins increased the inhibition of the bacterial enzyme by 220-fold, yet reduced the reaction with the human enzyme by 99.6 percent. Our success validated both the apparent substrate specificity of the M. tuberculosis proteasome and the structural model for proteasome inhibition by the syringolins."
The next step was to see whether the engineered compounds could indeed make bacteria more susceptible to nitric oxide, the chemical produced during the immune response. Sello's collaborators at the Weill Cornell Medicine added the engineered syringolins to cultures of M. tuberculosis in the presence and absence of a source of nitric oxide. As expected, they found that the bacteria treated with the compounds were highly susceptible to nitric oxide. In keeping with their weak inhibition of the human proteasome, the engineered syringolin did not inhibit the growth of human cell models.
"We were pleased to have engineered out the toxicity of the syringolins to human cells," Sello said. That suggested that an engineered syringolin could be safe in humans." Sello and his colleagues are hopeful that this initial round of testing could lay the groundwork for developing new drugs to treat tuberculosis. "We've only modified the syringolins in two ways," Sello said. "There are many other possibilities for structural modification that could improve potency and other pharmacological properties of the molecules. We can now see a long but feasible pathway towards the development of a novel therapeutic agent for tuberculosis."
Sello says it's plausible that a drug strategy like this one could be used alongside traditional antibiotics.
"One of the things that's clear in the treatment of tuberculosis is that combining drugs can be effective," he said. "So combining a blocker of the bacterium's defense against the immune system with a traditional antibiotic could be kind of a one-two punch."
More information: Kyle A. Totaro et al. Rational Design of Selective and Bioactive Inhibitors of theProteasome, ACS Infectious Diseases (2016). DOI: 10.1021/acsinfecdis.6b00172