Scientists add to theory about Huntington's mechanism
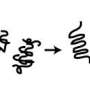
 encourages aggregation but is held in check by the proline C-terminus (blue) until polyglutamines reach a critical 36 repeats. Aggregation is suspected of causing Huntington's disease. Credit: Mingchen Chen/Rice University")
Rice University researchers are starting to understand how protein fragments influence the fiber aggregation suspected as a cause of Huntington's disease.
In their computer simulations, Rice bioscientist Peter Wolynes and graduate student Mingchen Chen show that the N-terminal sequence in huntingtin protein fragments encourages their aggregation into prefibrillar structures, while a C-terminal sequence made up of polyproline inhibits aggregation.
The models suggest their combined actions are part of the aggregation mechanism they described in a previous study about how repeats of polyglutamine genetically trigger Huntington's.
Wolynes said the discovery offers hope for drugs that could interfere with N-terminal binding and thereby stop aggregation early in the process. "Finding a target involves understanding molecular mechanisms and how things work at the atomic level, and we're adding to that part of the story," he said.
The research is detailed in the Proceedings of the National Academy of Sciences.
Huntington's is a hereditary disease caused by a mutation in the gene that expresses huntingtin proteins, which are common in human neurons and mostly harmless. The mutation involves a repeating chain of glutamines, which increases in length as genes are passed down through generations. Eventually, when the length surpasses a threshold, aggregation is triggered. Fibers typically begin to aggregate in Huntington's when these polyglutamine chains reach a critical length of 36 repeats. Longer chains can make the disease more severe and its onset earlier.
The Rice lab studies the molecular energy landscapes that allow proteins to fold into their functional shapes. This time, the researchers looked at the protein fragments that remain in a cell after the proteolysis - or breakdown - of large huntingtin proteins. These hairpin-like fragments contain polyglutamines capped on either end by an N-terminus and a C-terminus.
Wolynes and Chen found that the N- and C-termini are like children on either end of a see-saw: They must be in balance to stabilize fragments with long polyglutamine chains. If they are not, the fragments begin to form inclusion bodies, the aggregates found inside the cells of people with Huntington's.
"If you only had the N-terminus encouraging aggregation and you didn't have the polyproline at the other end, then everyone would get Huntington's disease," Wolynes said. "So the prolines are doing something clinically good, but we don't know why it has evolved that way."
Wolynes said the core process by which polyglutamines fold prematurely when their length is beyond the threshold remains the same as for peptides lacking the termini, "but it's modified quantitatively by the influence of the two ends." Some people may harbor huntingtin proteins with long polyglutamines that are prevented from causing disease by the presence of longer polyprolines, he said.
Altering the N-terminus may also halt progression of the disease. "What would happen if we change a single amino acid in the N-terminus? We can use our models to look into that," Wolynes said. "If there are people with 40 polyglutamines but who also have a mutation in the N-terminus head and they don't get Huntington's, that would be very interesting.
"But that's a very tricky thing to prove," he said. "If somebody doesn't have Huntington's disease, they usually don't come in and say, 'Check me out!' Still, this argues for finding drugs that modify N-terminal binding."
The researchers' simulations predicted that the critical length of fragments prone to aggregation is between 30 and 40 glutamines, which is "remarkably consistent with the critical length for disease onset," they wrote. "Before, we showed there's a critical length (of 30 glutamines) for this pre-folding transition into hairpin shapes, but that length was shorter than the actual disease onset length (of 36)," Wolynes said. "When you put on these two termini, the length of the protein moves up to the range where the disease onset actually occurs.
"This tends to point to aggregation itself as the cause of the illness, even though some have suggested that aggregation is a protective mechanism."
He said the results also underline the involvement of the cytoskeleton. Wolynes noted that the huntingtin protein is known to interact with at least four proteins linked to the cytoskeleton, the network of filaments that direct the transport around a cell. He said experiments have established that the cytoskeleton helps bring fragments together as they aggregate.
"I tend to think the weird involvement of the cytoskeleton is a clue, and the fact it's involved in other neural processes like long-term memory has a certain suspicious air about it," he said.
Wolynes said the Rice lab may probe proteolysis, in which enzymes cut larger proteins into fragments. "In almost all of the neurodegenerative diseases, including Alzheimer's and Huntington's, the aggregates that signify the presence of the disease are the result of proteolytic cleavage," he said. "Proteolysis seems to have evolved to cleave proteins at a specific point to do something functional, which suggests it's not merely an accident."
More information: Aggregation landscapes of Huntingtin exon 1 protein fragments and the critical repeat length for the onset of Huntington's disease, Proceedings of the National Academy of Sciences, www.pnas.org/cgi/doi/10.1073/pnas.1702237114