Researchers identify the genomic cause for Carey-Fineman-Ziter syndrome
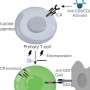
 fusing together to form muscle cells with more than one nucleus. The cascade is disrupted in Carey-Fineman-Ziter syndrome, because of a defect in the membrane protein, myomaker, which is required for cell-cell fusion. Credit: Darryl Leja, NHGRI")
An international team of researchers has identified genomic mutations for Carey-Fineman-Ziter (CFZS) syndrome, a very rare congenital myopathy (inherited muscle disorder) characterized by facial weakness, a small or retracted chin, a cleft palate and curvature of the spine (scoliosis), among other symptoms. The researchers determined that CFZS is caused by mutations in the gene MYMK that encodes for the protein myomaker. This protein is necessary for the fusion of muscle cells (myoblasts) into muscle fibers (myotubes) during the development of an embryo and the regeneration of muscle cells after injury. The study was published July 6, 2017, in Nature Communications.
"Advances in genomics technology and the power of team science have enabled us to identify the cause of this very rare disease 35 years after it was first described by Dr. John Carey and colleagues from the University of Utah," said National Institutes of Health Director Francis S. Collins, M.D., Ph.D., a co-author of the study. (See related story and video of Dr. Carey and a family affected by CFZS).
"This discovery will improve physicians' ability to diagnose this disease and offer families accurate genetic counseling and treatment," said Irini Manoli, M.D., Ph.D., co-lead author and a physician scientist and staff clinician in the Medical Genomics and Metabolic Genetics Branch at the National Human Genome Research Institute (NHGRI), part of NIH. People affected with CFZS have sometimes been misdiagnosed with Moebius syndrome, another very rare disorder characterized by facial paralysis.
Dr. Manoli said that uncovering that cell-cell fusion deficits can lead to congenital myopathies (inherited muscle disease) opens a new path of exploration for therapies for CFZS and other muscular diseases and tools for regenerating muscle. "In addition," she said, "this rare genetic syndrome provides novel insights into the effects of muscle development on craniofacial and skeletal bone formation."
In addition to NHGRI, study collaborators included researchers at the National Institute of Neurological Disorders and Stroke (NINDS), the NIH Clinical Center, the Boston Children's Hospital affiliated with Harvard University, Icahn School of Medicine at Mount Sinai in New York, the University of Utah in Salt Lake City and the University of Otago in Dunedin, New Zealand. The study resulted from a three-year effort by the Moebius Syndrome Research Consortium with support from NIH and the Moebius Syndrome Foundation.
The goal of the study was to learn more about the genetics and clinical characteristics of Moebius syndrome and other congenital facial weakness disorders. Toward this end, the consortium brought 63 people to the NIH Clinical Center affected with Moebius syndrome and other inherited facial weakness disorders, and their families for detailed multi-system evaluations, including brain and muscle imaging studies and muscle biopsies. The researchers collaborated through the Opportunities for Collaborative Research at the NIH Clinical Center, a new funding mechanism that encourages intramural and extramural researchers to work together at the NIH Clinical Center.
Researchers performed detailed phenotyping (identifying physical traits that are the result of a DNA sequence). They also employed the most up-to-date genomic tools, including exome sequencing of blood DNA in affected siblings from three unrelated families, as well as a muscle biopsy in one of the affected individuals. To identify the genomic mutations associated with CFZS, three laboratories—led separately by Elizabeth Engle, M.D., at the Boston Children's Hospital, Stephen Robertson, M.D., from the University of Otago and John Carey, M.D., at the University of Utah—analyzed exome sequence data from each of the three families. Among the genes harboring mutations identified in each family, only the gene MYMK was common to all three. A knockout mouse model (genomically altered mice that are bred to lack a specific gene) displayed a complete lack of muscle development, leading to early death of the newborn mice, making this gene a promising candidate for further studies.
Using CRISPR-Cas9 technology, a tool for editing DNA at precise locations, a team led by Silvio Alessandro Di Gioia, Ph.D., and Dr. Engle, generated zebrafish with a mutated mymk gene. Affected mutant zebrafish were smaller and had abnormal muscle development and jaw deformities, resembling the patient phenotype. The researchers then performed further functional studies to validate the severity of each of the genomic mutations.
The researchers were able to correct affected zebrafish's muscles by injecting the normal human MYMK gene product into the mutant fish. This success lends hope for restoring MYMK function in muscles as a treatment for CFZS and for reducing any potentially progressive features of this disorder.
Only eight people in the world have been diagnosed with CFZS with MYMK mutations, in part, because it hasn't been readily recognized. Now that researchers have identified the genomic cause underlying the syndrome, it can be added to the diagnostic gene panels for congenital myopathies. This will improve the speed and accuracy of diagnosis and add to the understanding of the spectrum of disease severity and outcome, Dr. Manoli said.
More information: Silvio Alessandro Di Gioia et al. A defect in myoblast fusion underlies Carey-Fineman-Ziter syndrome, Nature Communications (2017). DOI: 10.1038/ncomms16077