A math concept from the engineering world points to a way of making massive transcriptome studies more efficient

To most people, data compression refers to shrinking existing data—say from a song or picture's raw digital recording—by removing some data, but not so much as to render it unrecognizable (think MP3 or JPEG files). Now, biologists propose to bring a kind of data compression to molecular biology.
A Broad research team has proposed a new compression approach for gene expression (a.k.a. "transcriptomic") experiments, where the data volume per experiment is growing dramatically. Their approach—reported in Cell—leverages a mathematical framework called compressed sensing to collect a relatively small amount of data in the laboratory and mathematically "decompress" it. The result is a very close representation of a cell or tissue's full expression profile.
Engineers can use compressed sensing to reconstruct a signal's full content from just a few direct measurements, making data acquisition faster and cheaper. Some MRI machines, for example, use compressed sensing to scan patients more quickly.
To apply compressed sensing to transcriptomes, the team—led by graduate student Brian Cleary, postdoctoral researcher Le Cong, institute director Eric Lander, and core institute member and Klarman Cell Observatory (KCO) director Aviv Regev—relied on the fact that expression is:
- modular—cells do not express genes individually, but as sets in discrete programs—and
- sparse—each cell expresses only a limited number of modules at a time.

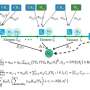
Taking advantage of these properties, the team thought it might be possible to construct transcriptomes using a few (up to 100-fold fewer than the number of genes) "composite" expression measurements (which sum multiple genes' weighted abundances into one measurement), instead of measuring every individual gene's expression. The researchers then developed an algorithm called BCS-SMAF (for Blind Compressed Sensing-Sparse Module Activity Factorization) that uses randomly collected composite measurements to identify active expression "modules."
The algorithm then reconstructs individual genes' expression within each module. Interesting, BCS-SMAF doesn't need prior information about which genes might constitute a module (e.g., cellular respiration genes or mTOR pathway genes).
In proof-of-concept experiments using various kinds of data (including simulated, published, or existing single-cell and bulk transcriptome data), the team found that BCS-SMAF produced composite-based expression profiles that closely fit the true profiles.
If validated in larger studies, the approach could provide deep insights into cells' active circuitry at greatly reduced experimental and computational costs—benefits that could spill over to other data-intensive biological fields such as proteomics or metabolomics.
More information: Brian Cleary et al. Efficient Generation of Transcriptomic Profiles by Random Composite Measurements, Cell (2017). DOI: 10.1016/j.cell.2017.10.023