Changing size of neurons could shed light on new treatments for motor neurone disease

New research published in The Journal of Physiology improves our understanding of how motor nerve cells (neurons) respond to motor neurone disease, which could help us identify new treatment options.
Motor neurone disease referred to as Amyotrophic Lateral Sclerosis (ALS) is associated with the death of motor nerve cells (neurons). It starts with the progressive loss of muscle function, followed by paralysis and ultimately death due to inability to breathe. Currently, there is no cure for ALS and no effective treatment to halt, or reverse, the progression of the disease. Most people with ALS die within 3 to 5 years from when symptoms first appear.
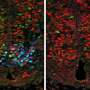
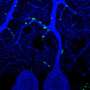
Previous studies in animal models of ALS have reported inconsistencies in the changes in the size of motor neurons. This new study is the first to show robust evidence that motor neurons change size over the course of disease progression and that, crucially, different types of neurons experience different changes. Specifically, the study shows that motor neuron types that are more vulnerable to the disease - that is, they die first - increase in size very early in the disease, before there are symptoms. Other motor neuron types that are more resistant to the disease (they die last) do not increase their size. These changes in the size of the motor neurons have a significant effect on their function and their fate as the diseases progresses.
The hope is that by understanding more about the mechanisms by which the neurons are changing size, it will be possible to identify and pursue new strategies for slowing or halting motor nerve cell death.
This research suggests motor neurons might alter their characteristics as a response to the disease in an attempt to compensate for loss of function. However these changes can lead to the neuron's early death. Furthermore the research supports the idea that the most vulnerable motor neurons undergo unique changes that might impact their ability to survive.
The research conducted by Wright State University involved identifying and measuring size changes of motor neuron types in a mouse model of familial ALS. The motor neurons were examined at every key stage of the disease to observe when and where these changes begin, and how they progress through the entirety of the disease. Specific antibodies were used as markers to bind to the structure of motor neurons so that they could be easily viewed under high-power microscopes, and a computer program performed the three-dimensional measurement of the size and shape of a motor neuron's cell body.
It is important to note that the research was carried out in only one mouse model which was the most aggressive mouse model of ALS. Future work should focus on other mouse models of ALS in order to determine how relevant these results are likely to translate in human patients.
Sherif M. Elbasiouny, the lead investigator on the research commented potential areas for further study:
"This research approach could be applicable not only to ALS, but also to other neurodegenerative diseases, such as Alzheimer's and Parkinson's diseases. This new understanding could help us to identify new therapeutic targets for improving motor neuron survival."
More information: S. Shekar Dukkipati et al, The vulnerability of spinal motoneurons and soma size plasticity in a mouse model of amyotrophic lateral sclerosis, The Journal of Physiology (2018). DOI: 10.1113/JP275498