Untangling brain neuron dysfunction in Parkinson's disease and dementia with Lewy bodies
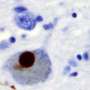
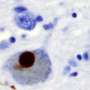
 of an intraneural Lewy-body in the Substantia nigra in Parkinson's disease. Credit: Wikipedia")
A decay of brain function is a hallmark of Parkinson's disease and dementia with Lewy bodies, or DLB. Specifically, cognitive dysfunction defines DLB, and nearly eight of every 10 Parkinson's patients develop dementia.
In both of these neuro-degenerative diseases, aggregates of misfolded alpha-synuclein protein develop in brain neurons, including the hippocampus, the region of the brain that plays a vital role in the formation of memories.
These aggregates eventually lead to cell death. However, knowledge of how the abnormal aggregates affect hippocampal neuron structure and function in Parkinson's and DLB before cell death is lacking.
Laura Volpicelli-Daley, Ph.D., assistant professor of neurology at the University of Alabama at Birmingham School of Medicine, and colleagues have now described changes in hippocampal neurons early after the pathogenic alpha-synuclein aggregates begin to appear. This understanding, combined with further exploration of the mechanisms underlying the neuronal changes, could point to novel therapeutic treatments to prevent or reverse neuronal defects and halt development of dementia.
"In Parkinson's disease, you can give levodopa to improve motor function; but there is nothing to stop the non-motor symptoms," Volpicelli-Daley said.
About 1 million Americans live with Parkinson's disease, and DLB is the second-most-common form of dementia in the elderly after Alzheimer's.
Volpicelli-Daley's study took advantage of a novel experimental model of alpha-synuclein aggregates in neurons developed by Volpicelli-Daley and colleagues at the University of Pennsylvania eight years ago. Fibrils resembling those found in PD and DLB brains can be made by putting synthetic alpha-synuclein in a test tube and shaking the solution for several days to allow alpha-synuclein to take on a fibrillary, pathologic conformation. These fibrils can be broken up and added to neurons that are grown in culture. The nerve cells take up some of the fibrils. Inside the cells, the fibrils attract the soluble alpha-synuclein that naturally is present in the cells to form the pathological, insoluble aggregates of alpha-synuclein.
These grow to form the typical inclusions emblematic of Parkinson's and DLB. As the pathologic alpha-synuclein inclusions continue to form, they increasingly impair neuronal excitability and connectivity, and eventually lead to neuron death.
In the present study, UAB researchers and a Yale University colleague looked at changes in mouse excitatory hippocampal neuronal function seven days after exposure to the fibrils, a time point before any of the neurons die. At seven days, alpha-synuclein inclusions are abundant in axons of the cells—the part of the nerve cell that sends a chemical signal to another nerve cell as part of nerve circuit functioning across the synapses between neurons. These neuronal circuits throughout the brain give rise to perception, action, thought, learning and memories.
The researchers found several significant changes in the hippocampal neurons. Formation of the pathological alpha-synuclein inclusions created multiple defects in both pre-synaptic and post-synaptic functions before neurodegeneration had begun.
"Something is clearly going on with the neurons before they die," Volpicelli-Daley said. "There is increased activity at the presynaptic terminal, the site of the neuron that releases chemicals called neurotransmitters. On the other hand, there is decreased activity post-synaptically, the site of the neighboring neuron where these released chemicals activate messenger systems. This may suggest that there is plasticity in the neurons, that is, the neurons are adapting to the increased activity."
"Over time, this abnormal activity may eventually lead to neuron death," she said. "The next step will be looking at how alpha-synuclein increases presynaptic activity and whether this is a loss of alpha-synuclein function in this neuron compartment or it is caused by formation of toxic alpha-synuclein aggregates."
"This is a groundbreaking study and one of the first to address critical and previously elusive questions regarding how toxic alpha-synuclein affects the structure and physiology of memory neurons," said Jeremy Herskowitz, Ph.D., assistant professor in the UAB Department of Neurology and the Patsy W. and Charles A. Collat Scholar of Neuroscience.
"Our team was able to move the field forward by applying a truly multidisciplinary approach," said Linda Overstreet-Wadiche, Ph.D., associate professor in the UAB Department of Neurobiology. "We combined anatomical, biochemical and functional assays to understand how inclusions alter neuronal function prior to cell death, and this approach yielded the unexpected finding that loss of postsynaptic structure was accompanied by enhanced presynaptic function."
Volpicelli-Daley, Herskowitz and Overstreet-Wadiche are co-senior authors of the study.
In detail, they found that synaptic activity in the absence of action potentials—specifically, miniature excitatory post-synaptic potentials—increased, even though there was a major reduction in spine density on the dendrites of the neurons. Electron microscopy also showed an increased number of docked presynaptic vesicles. Spontaneous synaptic activity driven by action potentials remained normal, but there was a major impairment in spontaneous influxes of calcium ions downstream of the synapse.
More information: Jessica M. Froula et al, α-Synuclein fibril-induced paradoxical structural and functional defects in hippocampal neurons, Acta Neuropathologica Communications (2018). DOI: 10.1186/s40478-018-0537-x