Calcium may play a role in the development of Parkinson's disease

Researchers have found that excess levels of calcium in brain cells may lead to the formation of toxic clusters that are the hallmark of Parkinson's disease.
The international team, led by the University of Cambridge, found that calcium can mediate the interaction between small membranous structures inside nerve endings, which are important for neuronal signalling in the brain, and alpha-synuclein, the protein associated with Parkinson's disease. Excess levels of either calcium or alpha-synuclein may be what starts the chain reaction that leads to the death of brain cells.
The findings, reported in the journal Nature Communications, represent another step towards understanding how and why people develop Parkinson's. According to the charity Parkinson's UK, one in every 350 adults in the UK - an estimated 145,000 in all - currently has the condition, but as yet it remains incurable.
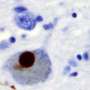
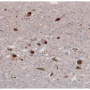
Parkinson's disease is one of a number of neurodegenerative diseases caused when naturally occurring proteins fold into the wrong shape and stick together with other proteins, eventually forming thin filament-like structures called amyloid fibrils. These amyloid deposits of aggregated alpha-synuclein, also known as Lewy bodies, are the sign of Parkinson's disease.
Curiously, it hasn't been clear until now what alpha-synuclein actually does in the cell: why it's there and what it's meant to do. It is implicated in various processes, such as the smooth flow of chemical signals in the brain and the movement of molecules in and out of nerve endings, but exactly how it behaves is unclear.
"Alpha-synuclein is a very small protein with very little structure, and it needs to interact with other proteins or structures in order to become functional, which has made it difficult to study," said senior author Dr Gabriele Kaminski Schierle from Cambridge's Department of Chemical Engineering and Biotechnology.
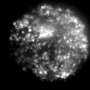
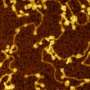
Thanks to super-resolution microscopy techniques, it is now possible to look inside cells to observe the behaviour of alpha-synuclein. To do so, Kaminski Schierle and her colleagues isolated synaptic vesicles, part of the nerve cells that store the neurotransmitters which send signals from one nerve cell to another.
In neurons, calcium plays a role in the release of neurotransmitters. The researchers observed that when calcium levels in the nerve cell increase, such as upon neuronal signalling, the alpha-synuclein binds to synaptic vesicles at multiple points causing the vesicles to come together. This may indicate that the normal role of alpha-synuclein is to help the chemical transmission of information across nerve cells.
"This is the first time we've seen that calcium influences the way alpha-synuclein interacts with synaptic vesicles," said Dr Janin Lautenschl?ger, the paper's first author. "We think that alpha-synuclein is almost like a calcium sensor. In the presence of calcium, it changes its structure and how it interacts with its environment, which is likely very important for its normal function."
"There is a fine balance of calcium and alpha-synuclein in the cell, and when there is too much of one or the other, the balance is tipped and aggregation begins, leading to Parkinson's disease," said co-first author Dr Amberley Stephens.
The imbalance can be caused by a genetic doubling of the amount of alpha-synuclein (gene duplication), by an age-related slowing of the breakdown of excess protein, by an increased level of calcium in neurons that are sensitive to Parkinson's, or an associated lack of calcium buffering capacity in these neurons.
Understanding the role of alpha-synuclein in physiological or pathological processes may aid in the development of new treatments for Parkinson's disease. One possibility is that drug candidates developed to block calcium, for use in heart disease for instance, might also have potential against Parkinson's disease.
More information: Janin Lautenschläger et al, C-terminal calcium binding of α-synuclein modulates synaptic vesicle interaction, Nature Communications (2018). DOI: 10.1038/s41467-018-03111-4