June 1, 2018 report
Nucleoside logic: Supply-side programming of the immune biocomputer

The immune system is host to a bewildering array of cell types. Traditionally, immunologists have classified cells in different states of activation according to the various interleukins, interferons and other cytokines they express or secrete. Unfortunately, this ever-sprawling matrix of cell markers has become a matrix of exceptions that is rapidly collapsing under its own weight.
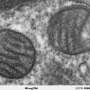
In this confusion, the specific metabolic state of any given immune cell class has emerged as a more reliable predictor of its status and fate. In particular, the condition of a cell's mitochondrial network is now proving to be the most fundamental classifier. Consider the monocyte cell lineage: These myeloid cells can be activated to become either inflammatory M1 macrophages, or tissue-healing M2 macrophages. Mitochondria in M1 macrophages run their own special version of the citric acid cycle which includes enzymatic blocks at two key points in the loop. The net effect of this is to create a hemicycle that extends from citrate to succinate that branches off into a mini urea cycle. This shunts metabolites into a pathway that generates nitric oxide used for host defenses.
Once thought to be an irreversible transition, M1 cells are now believed to be capable of re-differentiating to M2 cells and even vice-versa, making such traditional classifications problematic. A further case in point is the so-called giant multinucleated macrophage, which evolves from a local fusion of cells into 'supermacrophage.' As part of their respiratory burst attack, M1 macrophage mitochondria make special provisions for extra radical generation at complex I of the electron transport chain. This fortuitous formation of dangerous radicals can become a liability in the high oxygen environment of the bloodstream.
Consider the T cell lineage: Any given 'naive' T cell is capable of undergoing rapid clonal expansion to meet immunological needs as effector or helper T cells. This proliferative phase bears some parallel to cancer cells in a rapidly growing tumor in that a metabolic shift to glycolysis is required to generate precursors for cell division. While most of these cells will expire when their job is done, some of them will escape apoptosis to persist indefinitely as memory cells. These particular cells are perhaps more reflective of post-mitotic neurons, or perhaps even the syncytial multinucleated cells of muscle. This transition depends on proliferation of mtDNA within the T cell mitochondrial network to establish what is known in the business as 'reserve respiratory capacity.'
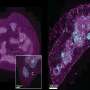
But what might predict, or rather limit, the status of the mitochondrial network? During the early stages of the T cell life cycle, thymocyctes prepare to enter the oxygen-rich bloodstream by removing surplus radical-generating mitochondria via autophagy and lysosomal reprocessing. A paper just published in Cell Reports neatly ties these processes together by showing that the mitochondrial set point is adjusted across the T cell life cycle by controlling access to nucleotide building blocks. More specifically, the authors show that the limiting conduction of nucleosides (which are precursors to nucleotides) across organelle membranes by the blocking ENT3 transporter disrupts lysosomes and creates an excess of mitochondria.
The local nucleotide ecosystem that predominates within any cell or tissue can be considered a matrix unto itself. As an example, we might consider that each row and column of such a transition matrix will need to have entries not just for the standard bases (A,G,C,T,U), but also all the intermediary bases and breakdown products which come into play (like inosine and xanthine). Additionally, we would also need to include the successive stages of processing of each nucleobase through the nucleoside form, and then the monophosphate, diphosphate, and triphosphate nucleotide forms, and ultimately generation of the deoxynucleotide for the DNA. Each cell type will have only a partial ability to make each transition represented in such a matrix because each cell only possesses a subset of the all the corresponding enzymes available in the genome.
T cells, and for that matter many other immune cells, lack much of the normal de-novo purine and pyrimidine synthesis enzymes in their nuclei and their mitochondria. These low level inputs to the matrix must therefore be shared from other sources via transporters like ENT3, or obtained by nucleotide salvage pathways. The immune system is an assembly of nucleotide specialists where each cell type contributes special enzymes to process shared nucleotide pools. In so doing the whole defensive network is able to finesse attacks to rapidly multiply invaders of many different kinds. When there are hits to any component of the nucleotide matrix, the result is typically a very specific and debilitating medical condition.
For example, those with point variants in ENT3 mutations are invariably awarded with any of a wide variety of heterogeneous hereditary diseases. This includes pigmentary hypertrichosis, H syndrome, non-autoimmune insulin-dependent diabetes mellitus syndromes, and Rosai-Dorfman disease. ENT3 is one of 4 genes in the 'equilibrative nucleotide transporter' family. It is the one found predominantly on mitochondria and lysosomes, and isalso upregulated in many common cancers. The other three ENT versions have been implicated in the bidirectional transport of nucleoside analogues (like AZT) currently used in different chemotherapies. Other genes involved in nucleotide salvage and distribution can cause similarly unique problems for T cells. Deficiencies in deoxycytidine kinase, for example, creates replication stress and genetic damage by interfering with DNA repair.
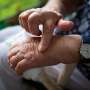
To get a richer appreciation for the role of mitochondria and T cell function in immunity it we take a more specific example. Poison ivy initiates a massive immune reaction when urushiol oil gets on our skin and becomes oxidized to an electrophilic quinone derivative. This quinone then acts as a hapten that attaches to specific amino acids in proteins like the keratin made inside keratinocytes. Although it is not yet known why these conjugated quinones are seen as foreign enemies, we do know that longer and more unsaturated the hydrophobic tail of the quinone is, the greater the response.
These immunogenic quinone epitopes are processed by antigen-presenting Langerhans cells in the dermis which then bind to receptors on T cells. Once activated, these T cells in turn recruit macrophages to attack the offending locations on the skin. Normally, extrinsic (exogenous) antigens are endocytosed into the lysosome compartment and bound to class II MHC molecules that get recognized by CD4+ T cells. Endogenous proteins, on the other hand, are degraded into peptides approximately nine amino acid length in the endoplasmic reticulum and then get bound to class I MHC molecules for presentation to CD8+ T cells.
This tidy picture can't fully explain the full poison ivy reaction because both CD4+ and CD8+ pathways are engaged at different levels. Urushiol-derived quinones clearly modify endogenous proteins and therefore are effective at activating CD8+cells. However, a separate CD4+ activation to urushiol has been found that first potentiates and then dampens the overall immune response.
At each step, mitochondria have been found to play the decisive role. They generate the initial oxidative burst that converts urushiol to a quinone. Next, the hydrophobic chains of the quinone are shortened inside the mitochondrial matrix by beta-oxidation in order to be able to bind to the MHC molecules. Control of the mitochondrial network then establishes activation and later immunological memory in T cells, ultimately activating death squad recruitment in macrophages.
More information: Chin-Wen Wei et al. Equilibrative Nucleoside Transporter 3 Regulates T Cell Homeostasis by Coordinating Lysosomal Function with Nucleoside Availability, Cell Reports (2018). DOI: 10.1016/j.celrep.2018.04.077
© 2018 Medical Xpress