Scientists eradicate cancer cells through dual targeting of DNA repair mechanisms
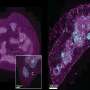
 are color-coded in pink, orange, purple and yellow. Credit: NHGRI")
Proteins commonly known as BRCA—short for BReast CAncer susceptibility gene- serve a critical role in cellular DNA repair, but when mutated they allow genetic errors to replicate, facilitating cancer development. If the BRCA repair system is disabled in cancer cells, the cells simply turn to backup repair mechanisms and adapt to alternative repair pathways, a survival mode that also underlies their ability to evade targeted drug therapies.
Now, new research by scientists at the Lewis Katz School of Medicine at Temple University (LKSOM) shows that it is possible to eliminate at least two backup repair mechanisms at the same time using two targeted therapies. The strategy effectively narrows down the number of secondary repair pathways available, helping to ensure cancer cell eradication.
The novel approach, described June 12 in a paper published in Cell Reports, is named dual synthetic lethality, so-called because cancer cell death is induced by two drugs targeting distinct DNA repair pathways at the same time. "Cancers cells have multiple ways of protecting themselves from death," explained senior investigator Tomasz Skorski, MD, Ph.D., Professor of Microbiology and Immunology and Associate Professor at the Fels Institute for Cancer Research and Molecular Biology at LKSOM.
Tumor cells that carry BRCA-deficiency mutations frequently rely on DNA repair mediated by a protein known as PARP1. Clinically, PARP1 inhibition can significantly improve progression-free survival in patients with cancers involving hereditary BRCA mutations.
Over time, however, the effectiveness of PARP1 inhibition declines, and cancer relapses. According to Dr. Skorski, this happens because targeting one pathway is not enough. "The tumor cells eventually escape PARP1 inhibition by activating another backup to the BRCA-mediated repair pathway," he said. "Our previous work had suggested that RAD52-dependent pathways are a likely escape route, which led us to see whether simultaneous inhibition of both PARP1 and RAD52 could trigger more effective lethality."
Dr. Skorski's team first tested their idea in a series of experiments in BRCA-deficient solid tumor and leukemia cell lines in vitro. Using an experimental RAD52 inhibitor, they were able to show that RAD52 blockade enhanced synthetic lethal effects in cells treated with the PARP inhibitor olaparib, a drug approved by the Food and Drug Administration. For all tumor types, the dual treatment completely eradicated BRCA-deficient tumor cells.
The simultaneous targeting strategy was then tested against BRCA-deficient tumors in mice, in which the researchers recapitulated the effects of BRCA deficiency in breast cancer and in acute and chronic myelogenous leukemia. They found that dual inhibition exerted strong effects against tumors in vivo, showing greater activity than either agent alone.
Importantly, the teams' experiments showed, both in vitro and in vivo, that normal cells, with normal BRCA activity, are unaffected by the dual treatment. "Normal cells continue to use BRCA-mediated repair as their primary DNA repair pathway and do not rely heavily on PARP1 or RAD52," explained Katherine Sullivan-Reed, a graduate student in Dr. Skorski's research laboratory and first author on the new paper. Leaving normal cells relatively unharmed helps limit the severity of side effects patients may experience with targeted cancer therapy.
The new research provides a foundation for pursuing the development of a clinically viable drug to inhibit RAD52, as well as the development of a screening test specifically for patients who carry tumors displaying BRCA-deficiency.
In the near-term, Dr. Skorski's team also plans to continue investigating alternative DNA repair pathways, which are essential for tumor cells but expendable in normal cells. "One of our primary goals is to find additional targets to kill cancer cells, without increasing toxicity," he added.
More information: Cell Reports (2018). DOI: 10.1016/j.celrep.2018.05.034