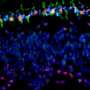
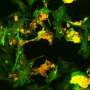
Team identifies developmental stage for no. 1 eye tumor in children
 and areas of RB protein loss (red). Credit: Hardeep Singh, PhD, of Children's Hospital Los Angeles")
Investigators at Children's Hospital Los Angeles have been able to pinpoint the exact stage of development of the human retina, when cells can grow out of control and form cancer-like masses. The finding could open the door for future interventions in retinoblastoma (RB), a tumor of the retina that affects children under five years of age.
The study is a continuation of research supported by a grant from the National Cancer Institute and was published online Sept. 13 in the prestigious journal PNAS, Proceedings of the National Academy of Sciences.
The investigation represents the first of its kind by identifying the phase of human retinal development when specific cells—called cone precursors—may turn cancerous.
"Understanding this phase of development and what goes wrong can help us find ways to intervene and eventually prevent retinoblastoma," said David Cobrinik, MD, Ph.D., of The Vision Center at Children's Hospital Los Angeles.
Although rare, retinoblastoma is the most common malignant tumor of the eye in children and can lead to devastating vision loss. CHLA is considered a world leader in the research and treatment of the disease, which can be fatal if not diagnosed early.
In a prior breakthrough in 2014 that led to this study, the CHLA researchers identified cone precursor cells as the cell-of-origin of retinoblastoma. Cone cells, found in the retina, are responsible for color vision.
Following up on the 2014 discovery with the current study, the team found that at a specific point in their maturation, human cone precursors cells can enter the cell cycle—this is a series of events leading to their division. The cells then begin to proliferate and form pre-malignant lesions that can develop into rapidly growing retinoblastoma-like masses. The maturing cone precursors enter the cell cycle in response to the inactivation of the RB1 tumor suppressor gene and loss of functional RB protein, which regulates cell growth and keeps cone precursor cells from dividing.
"We suspect that the maturing cone precursors are wired in a way that causes them to become cancer cells in response to loss of the RB protein," said Cobrinik, an investigator with The Saban Research Institute of CHLA and associate professor of Ophthalmology at the Keck School of Medicine at the University of Southern California.
In another key finding, the investigators compared the developmental process of the human eye to a traditional mouse model. Lead author and postdoctoral research fellow Hardeep Singh, Ph.D., found that developmental stage-specific proliferation and formation of retinoblastoma occurred in RB-deficient human cone precursors but not in mouse precursors. The animal models failed to replicate the genetic, cellular, and developmental features of human retinal cells. This finding calls into question the accuracy of certain animal retinoblastoma models.
An alternative way to study the condition could involve induced pluripotent stem cells, said Cobrinik. These can be generated directly from adult cells and are another subject of investigation in his laboratory.
Retinoblastoma was one of the first tumors to have its genetic cause identified. RB1 tumor suppressor gene mutations were identified at CHLA and other institutions about 30 years ago. Since that time, much has been learned about how RB1 mutations initiate retinoblastoma tumors.
"Given the current state of genomic analyses," said Cobrinik, "we can look forward to a time when we will be able to test for mutations in RB1 as well as other disease-associated genes and provide disease-preventing interventions."
More information: Hardeep P. Singh et al, Developmental stage-specific proliferation and retinoblastoma genesis in RB-deficient human but not mouse cone precursors, Proceedings of the National Academy of Sciences (2018). DOI: 10.1073/pnas.1808903115