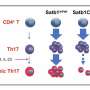
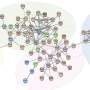
A new way to map cell regulatory networks
 that regulate the function of T Helper 17 (TH17) cells, an immune cell type that defends against bacterial and fungal infections but also can lead to autoimmune diseases when malfunctioning. In this image, bolded gene names correspond to transcription factors (TFs). Connections between TFs and genes are colored red for activation or blue for inhibition of gene expression. Credit: Cincinnati Children's")
Faster results. Lower costs. Fewer cells.
A new mathematical method developed by researchers at Cincinnati Children's and New York University may soon make it much easier to conduct more of the complex data analysis needed to drive advances in the exploding field of personalized medicine.
Proof-of-principle results for the method are reported this month in Genome Research. The co-authors say they were able to use vastly less biological material to reproduce—and even expand upon—an expensive previous effort to identify a network of protein transcription factors (TFs) that regulate the function of T Helper 17 (TH17) cells, an immune cell type that defends against bacterial and fungal infections but also can lead to autoimmune diseases when malfunctioning.
The learnings regarding TH17 cells are significant in their own right, researchers say. The data adds several TFs to the known transcriptional regulatory network (TRN) that controls how TH17 cells behave. This, in years to come, could lead to better treatments for autoimmune diseases like multiple sclerosis, psoriasis and inflammatory bowel disease.
However, this discovery's potential impact may reach much further.
"This method could be applied to any cell type, and could provide new information on how targeted cell types could be controlled in a wide range of diseases," says Emily Miraldi, Ph.D., a computational biologist at Cincinnati Children's and lead author on the new study.
In years to come, detailed knowledge about the TRNs controlling specific cell types could help researchers find precise ways to ramp up immune system responses to attack cancer tumors or tamp down responses to treat a variety of immune diseases, Miraldi says.
From 10-million-cell samples to as few as 500
Using current methods, researchers have learned much about how gene expression can differ among healthy and unhealthy people. But learning how to safely influence gene expression to improve health involves drilling deeper into how TRNs orchestrate behavior in specific cell types, locations and conditions.
In many diseases, especially rare ones, it can be difficult to impossible to gather enough cells of interest to conduct such analyses, Miraldi says.
For example, detailing the initial regulatory network for TH17 cells relied on data from chromatin immunoprecipitation sequencing (ChIP-seq), a breakthrough technology introduced about a decade ago.
But that tool requires as many as 10 million cells to work. The methods used by Miraldi and colleagues produced similar results with 100 times fewer cells. In fact, in some cases, data could potentially be obtained from samples with as few as 500 cells.
In the new approach, the research team used an improved version of a gene expression modeling algorithm called "Inferelator" that integrates data from a newer technique called assay for transposase-accessible chromatin (ATAC-seq).
ATAC-seq zeroes in only on the portions of chromatin—the long, coiled strands of DNA within each cell—that are "open" for transcription factors to bind. "ATAC-seq is a possible alternative to multiple ChIP-seq experiments. I call it a poor man's ChIP-seq," Miraldi says.
Analysis of ATAC-seq information required several other tools as well, including a massive database (CisBP) of transcription factor binding preferences for thousands of transcription factors. The CisBP database was assembled by a team led by Cincinnati Children's computational biologist Matthew Weirauch, Ph.D.
If the upgraded Inferelator algorithm proves as useful as hoped, it could help reduce some of the need for animal research. Instead of spending months raising hundreds of carefully gene-modified mice to produce an adequate supply of a targeted cell type, it may become possible to obtain valid results using cell samples from as few as two or three mice, Miraldi says.
More study needed to apply and improve the new method
Miraldi and colleagues have already begun pursuing two lines of study related to this discovery.
One project applies the new method to construct TRNs for intestinal immune cells in vivo.
Another project seeks to refine the new method so that it can be applied to even smaller samples, such as those isolated from human patients using single-cell genomics.
More information: Emily R. Miraldi et al, Leveraging chromatin accessibility for transcriptional regulatory network inference in T Helper 17 Cells, Genome Research (2019). DOI: 10.1101/gr.238253.118