Early research shows a way to develop effective therapies against pancreatic cancer

Pancreatic ductal adenocarcinoma (PDAC), the most common form of pancreatic cancer, is the fifth most common cause of death from cancer in the UK and the third in the United States. Deaths from PDAC outnumber those from breast cancer despite the significant difference in incidence rates.
Late diagnosis and ineffective treatments are the most important reasons for these bleak statistics.
PDAC is an aggressive and difficult malignancy to treat. Until now, the only chance for cure is the complete surgical removing of the tumour. Unfortunately, because PDAC is usually asymptomatic, 80% to 90% of patients have disease that is surgically incurable by the time it is diagnosed. PDAC thus remains one of the main biomedical challenges today due to its low survival rate—about 5% of patients are still alive five years after diagnosis.
However, in recent decades a number of studies have shed light on the molecular mechanisms responsible for the initiation and progression of PDAC. Our recent research has shown that progress toward a cure is possible.
Ineffective treatments
The molecular mechanisms responsible for pancreatic cancer are complex, which is why recent advances in personalised medicine and immunotherapy (which helps the immune system fight cancer) have failed to improve the treatment of pancreatic cancer. This is mainly due to two characteristics:
- 95% of these tumours are caused by mutations in KRAS oncogenes. Oncogens are genes that once mutated are capable of inducing the transformation of a normal cell into a cancerous cell. KRAS is a gene that acts as an on/off switch. Normally, it controls cell proliferation, but when it is mutated, the cells starts to grow uncontrollably and proliferate—a hallmark of a cancer cells. So far, KRAS oncogenes have not be able to be targeted by drugs.
- PDACs are surrounded by abundant desmoplastic stroma, a fibrous connective tissue that grows around some tumour types. In the case of PDAC, it forms a barrier that prevents cytotoxic T lymphocytes (a type of cell that can recognise and attack tumour cells) from reaching the inside of the tumour mass and killing its cells. This renders immunotherapy treatments useless.
For these reasons, PDAC continues to be treated with cytotoxic drugs, which destroy cancerous cells but can also destroy healthy ones. Options include Demcitabine, approved in 1997, and Nab-paclitaxel, a new paclitaxel-based formulation. Even if such a treatment is an option, it only extend the patients' lives a few weeks, a marginal improvement at best.
In recent decades, however, a number of studies have shed light regarding the molecular mechanisms responsible for the initiation and progression of PDAC.
Today we know that most of these tumours are caused by mutations in the KRAS oncogene. They lead to benign alterations that cause additional mutations in a range of tumour-suppressor genes, which usually repair DNA mistakes, slow down cellular division or tell cells when to die. When they are mutated, cells can grow out of control, and in this context progress to malignant PDAC. While this process is relatively well known, it has not had an immediate impact on the development of new and more effective treatments.

In search of new strategies
Multiple strategies are currently being studied in an attempt to inhibit the growth of these tumours by blocking the growth of either the tumour cells or their surrounding "shielding" desmoplastic stroma. In our laboratory, we focused on blocking the signalling pathways that mediate the oncogenic activity of the initiating KRAS oncogenes.
A decade ago our lab decided to use genetically engineered mouse-tumour models capable of reproducing the natural history of human PDAC in order to analyse the therapeutic potential of the main components of the KRAS signalling pathways. These studies have unveiled the reason why the drugs tested so far have intolerable toxic effects, with mice dying within several weeks: they target some proteins that are essential for the dynamic state of equilibrium that is the condition of optimal functioning of the cells (a state called normal homeostasis).
These crucial proteins are mainly kinases, enzymes that are able to modify other molecules by chemically adding phosphate groups to them, altering their functions. Kinases play a critical and complex role in regulating cellular signalling, and they orchestrate multiple processes (hormone response, cell division…). These results might explain why the inhibitors of KRAS signalling tested so far have failed in clinical trials. On the other hand, the removal of other signalling kinases did not have toxic side effects, but also had no impact on tumour development.
Of the more than fifteen kinases involved in the transmission of signals from the KRAS oncogene, only three displayed significant therapeutic benefits without causing unacceptable side effects. These are: RAF1, the epidermal growth factor receptor (EGFR) and CDK4.
It works! (in mice)
In initial studies, we observed that the elimination (via genetic manipulation) of the expression of some of these three kinases prevented the onset of PDAC caused by the KRAS oncogene. However, its elimination in animals with advanced tumours had no significant therapeutic effects. These results caused us to question whether it would be possible to eliminate more than one kinase simultaneously without increasing the toxic effects.
As described in our recent work published in the journal Cancer Cell, the elimination of RAF1 and EGFR expression induced the complete regression of advanced PDACs in 50% of the mice. We are currently studying whether we can increase this percentage by also eliminating CDK4.
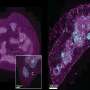
The analysis of the pancreas of animals in which we were no longer able to observe tumours by imaging techniques revealed the complete absence of lesions in two of them. Two animals showed some desmoplastic tissue residue with abnormal ducts, probably residual scarring from the tumour. The other mice had tumour micro-masses of one-thousandth the size of the original tumour. The study of these revealed the presence of tumour cells, in which the expression of the two targets, EGFR and RAF1, had not been completely eliminated, a common technical problem in this type of studies.
Significantly, these results were observed not only in mice. The inhibition of the expression of these two proteins in cells derived from nine out of ten human PDACs were also capable of blocking their proliferation in vivo when transplanted into immunosuppressed mice as well as in in vitro cultures.
Now what?
While these results have only been observed in a subset of mice for now, their importance lies in the fact that it is the first time that it has been possible to completely eliminate advanced PDAC tumours by eliminating a pharmacologically directed target.
These observations are clearly important for the development of treatments based on the inhibition of RAF1 and EGFR, but they only represent a first step on a long, hard road ahead.
First, it is important to identify the differences between the PDACs that respond to the combined elimination of RAF1 and EGFR and those that are resistant. As described in our work, the analysis of these two tumour types revealed that they are not active in the same way—more than 2,000 genes are expressed differently.
Identifying additional targets in resistant tumours that do not increase treatment toxicity is not going to be an easy task.
To continue our tests with genetically engineered mice, the immediate but no less difficult task is the development of specific RAF1 inhibitors. Indeed, we only currently have potent drugs against the second target, EGFR. In principle, there are four possible approaches:
- Generate selective inhibitors for its kinase activity.
- Generate inhibitors for its binding to the KRAS oncogene.
- Generate inhibitors for its interaction with effector targets that transmit oncogenic signalling mediated by RAF1.
- Degrade the RAF1 protein with drugs.
Designing inhibitors of the RAF1 kinase activity would seem to be the most affordable option, given the experience of the pharmaceutical industry in designing this molecule type.
The problem resides in the fact that there are two other kinases of the same family, ARAF and BRAF, whose catalytic centres (the "active core" of the enzymes) are nearly identical. RAF1 kinase inhibitors are also targeting these other kinases, which causes collateral damage. The ones tested to date have caused high toxicities and the clinical trials had to be stopped.
Continuing to develop effective molecules that are capable of blocking RAF1 activity in patients with PDAC will not be easy. It will surely take more time than we hope, but at least a roadmap has already been outlined that shows us how to keep moving forward.
This article is republished from The Conversation under a Creative Commons license. Read the original article.![]()















{kind=link}