A kinase identified as possible target to treat heart failure

An unexplored kinase in heart muscle cells may be a good target to treat heart failure, a disease that is only incrementally delayed by existing therapies. Failing human hearts showed reduced amounts of this kinase, and preclinical experiments showed that restoring the amount of this kinase in a kinase-depleted mouse model rescued the animal from heart failure, according to a study published in the journal Circulation and led by University of Alabama at Birmingham researcher Hind Lal, Ph.D.
Heart failure, especially in the months or years after heart attacks, is a prevalent, costly and growing cause of morbidity and mortality worldwide.
The signaling kinase is HIPK2. While several hundred protein kinases have been identified in heart muscle cells of failing human hearts, study has so far neglected the majority of highly expressed heart kinases, including HIPK2.
"Our study identifies HIPK2 as a novel regulator of heart failure progression," said Lal, an associate professor of medicine in the UAB Division of Cardiovascular Disease.
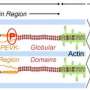
Regulator kinases like HIPK2 are the enzymes that can relay a signal from the surface of the cell—working through a cascade of kinases—to the cell nucleus, where the signal alters expression of genes in the chromosome.
To identify HIPK2 as a good candidate for study, Lal and colleagues took a bioinformatics approach. They first identified genes that were differentially expressed in failing mouse hearts, and next used three different algorithms to postulate the transcription factors responsible for those expression changes. Finally, they linked those transcription factors with potential upstream kinase regulators.
"Among those candidates, we identified a potential modulator of heart failure—HIPK2, a kinase whose role in cardiac biology has never been studied before," Lal said. "To determine the role of HIPK2 in failing human hearts, we examined the expression of HIPK2 in heart tissue from patients with end-stage ischemic cardiomyopathy. The expression of HIPK2 was dramatically decreased in failing hearts compared with normal human hearts."
That set the stage for deeper study.
The major findings—found in experiments with mouse models and rat heart-muscle cells—were that deletion of HIPK2 from heart muscle cells, known as cardiomyocytes, led to progressive deterioration of heart function with age. Younger mice had normal heart function. At the molecular level, researchers showed that the HIPK2 deficiency caused enhanced programmed cell death, or apoptosis, of the cardiomyocytes in heart muscle. Mechanistically, the HIPK2 deficiency and apoptosis signaling acted via decreased ERK1/2 kinase phosphorylation.
Lal and colleagues speculate that cardiac-specific restoration of HIPK2 in failing human hearts may be a way to slow disease progression.
Details of the study
The researchers generated mice with HIPK2 knocked-out in cardiomyocyte cells. At 2 months of age, those knockout mice that lacked heart-muscle HIPK2 had the same level of heart function as controls. However, at 3 months they showed significantly decreased heart function—lower ejection fraction and a fractional shortening—and also had significant elevation of the heart failure markers NPPA and NPPB.
The knockout mice did not show changes in fibrosis or calcium handling compared with controls. At 8 months of age, the heart failure in knockout mice was much worse, and the hearts showed remodeling and enlargement.
The researchers also studied heterozygous mice, where one HIPK2 gene was deleted in the cardiomyocytes and one was left intact. These haploinsufficient mice had comparable heart function as the controls at 3 months, but by 6 months, their heart function was significantly reduced, and the mice showed left ventricular dilation and thinning of the heart muscle wall.
"Thus, these findings indicated a direct dose-dependent relationship between the level of cardiomyocyte HIPK2 expression and cardiac function," Lal said.
Researchers found that phosphorylation of the kinases ERK1/2 was significantly decreased in the heterozygous mouse hearts, suggesting that HIPK2 acted through ERK signaling.
In experiments with neonatal rat ventricular cardiomyocytes, they found that knockdown of HIPK2 significantly decreased both ERK1 and ERK2 phosphorylation; in contrast, overexpression of HIPK2 led to significant elevation of ERK1/2 phosphorylation. Furthermore, this overexpression depended on HIPK2 retaining an intact kinase domain.
It is well-known that ERK signaling protects the heart from stress-induced apoptosis, which is a key driver of cardiac remodeling and heart failure. HIPK2 is also known to be a key regulator of apoptosis as well. Therefore, Lal and colleagues looked for apoptosis in the HIPK2 knockout mice. They found a significant elevation of the pro-apoptotic molecule BAX in heart muscle, and histology showed a clear elevation of TUNEL-positive nuclei in heart muscle tissue, a sign of cells undergoing apoptosis.
To test the hypothesis that impaired ERK signaling in the HIPK2-knockout hearts was the primary driver of cardiac dysfunction, the researchers did a rescue experiment. They used gene therapy to add overexpression of the kinase MEK1 to the knockout cardiomyocytes. MEK1 is known to act upstream of ERK in the kinase signaling cascade. This addition not only restored ERK phosphorylation in the knockout mice; it also significantly improved the ejection fraction and fractional shortening of the mouse hearts, and it decreased apoptosis of the knockout cardiomyocytes.
More information: Yuanjun Guo et al, Cardiomyocyte HIPK2 Maintains Basal Cardiac Function via ERK Signaling, Circulation (2019). DOI: 10.1161/CIRCULATIONAHA.119.040740