Uncuffing nitric oxide production: Beta-arrestin2 complexes regulate NO levels
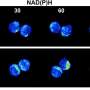
 localizes with eNOS (purple) in healthy cells (top panel); however, upon injury (lower panel), this localization is lost. Credit: Dr. Don Rockey.")
The liver filters blood, detoxifies chemicals and metabolizes drugs. Hepatitis, alcohol and primary liver diseases are common sources of damage in the liver and can result in scarring, known as fibrosis. Long-term fibrosis can lead to thicker liver tissue which in turn leads to other issues like increased blood pressure in the liver. Known as portal hypertension, high blood pressure in the liver is often unrecognized, and can be lethal.
Researchers at the Medical University of South Carolina (MUSC) who study liver disease have dissected one mechanism that regulates the activity of endothelial nitric oxide synthase (eNOS), an enzyme that produces nitric oxide (NO). Their results, published recently in the Proceedings of the National Academy of Sciences, showed that beta-arrestin2 (beta-Arr2) acts as a scaffolding protein to activate eNOS, leading to increased NO synthesis. Upon liver damage, levels of beta-Arr2 are decreased in epithelial cells causing a disruption in NO synthesis and an increase in portal hypertension. This research shows that beta-Arr2 is a potential therapeutic target.
"The molecular mechanisms that regulate eNOS function are complex and likely vary in normalcy and disease," said Don C. Rockey, M.D., professor and chair of the Department of Medicine at MUSC, who studies liver fibrosis and portal hypertension.
The Rockey Lab focuses on understanding the underlying mechanisms of portal hypertension. Liver sinusoidal endothelial cells (SECs) play a critical role in liver homeostasis. SECs control blood pressure and flow through the production of NO, which is produced by eNOS. As a signaling molecule, beta-Arr2 has been suggested to influence eNOS, but how these proteins interact to influence NO production was unclear.
The current work delineated the role of beta-Arr2 in regulating eNOS activity. In normal, healthy SECs, beta-Arr2 activated eNOS. In contrast, injured SECs showed a significant reduction in beta-Arr2 levels and decreased eNOS activity. Interestingly, overexpression of beta-Arr2 enhanced NO production in both normal and injured SECs. Furthermore, mice that lacked beta-Arr2 had a significant reduction in sinusoid area, highlighting the physiological effects of reduced eNOS activity. Together, these data show that beta-Arr2 is a critical regulator eNOS activity.
Previous work from the Rockey lab has shown that GPCR kinase-interacting protein-1 (GIT-1) is a potent activator of eNOS. Examination of GIT-1 localization revealed that GIT-1, beta-Arr2 and eNOS all localize together in SECs. Importantly, the loss of beta-Arr2 prevented the activation of GIT-1 and the subsequent activation of eNOS. Therefore, beta-Arr2 acts as a scaffolding protein to help activate GIT-1 and in turn activate eNOS.
In summary, healthy SECs produce NO by stimulating beta-Arr2, which then forms a signalosome, an active protein complex that helps transduce cell signals, to activate GIT-1. Active GIT-1 in turn activates eNOS to produce nitric oxide. In contrast, during liver injury, levels of beta-Arr2 are reduced and this pathway is perturbed.
"The big message is that eNOS signaling is complex and is controlled by multiple mechanisms and is dysregulated after injury to endothelial cells," said Rockey.
As a central component of the eNOS signaling axis, beta-Arr2 has the potential to be a novel therapeutic target for the restoration of SEC function. However, before a potential therapeutic makes it to the clinic, there are still several questions remaining about the biology of beta-Arr2. As a signaling scaffold that activates eNOS, it is important to determine what other proteins interact with beta-Arr2. Additionally, the Rockey lab is pursing the mechanism(s) that influence beta-Arr2's localization and/or activity.
More information: Songling Liu et al, β-Arrestin2 is a critical component of the GPCR–eNOS signalosome, Proceedings of the National Academy of Sciences (2020). DOI: 10.1073/pnas.1922608117