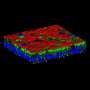
Restoring airway integrity in cystic fibrosis patients
. Credit: UNIGE - Laboratory of Prof. Marc Chanson")
Cystic fibrosis is a rare genetic disease which can cause very serious symptoms. In particular, patients suffer from chronic bacterial infections that can lead to respiratory failure. It is caused by mutations in the CFTR gene, which regulates water movement across the cell membrane. Consequently, when mucus quality is altered, it is no longer capable of capturing undesirable bacteria and expelling them. Using a model reproducing a respiratory epithelium—a protective tissue composed of a monolayer of cells—teams from the University of Geneva (UNIGE) have discovered that a simple film of liquid is sufficient to restore the airways' seal and reduce the risk of bacterial infection. These results, published in a special issue of the journal Cells, open the way to new therapies based on mucus hydration. A promising alternative to current therapies that are often not widely enough effective.
Despite recent therapeutic advances, people with cystic fibrosis—one in every 2,500 births in Europe—have a life expectancy of no more than 46 years and altered quality of life. The disease is caused by one or more mutations in the CFTR gene, which affects the proper functioning of an essential protective barrier. The epithelial cells that line the airways are usually sealed together and thus protect the airways from bacterial colonization. They are also lined with a fluid, a slippery mucus that traps unwanted germs and carries them away. When the CFTR protein is altered, the junctions between the cells loosen and the dehydrated mucus tends to stagnate, both of which promote the development of respiratory infections.
"While it was already known that mucus hydration and the presence of sufficiently tight junctions preserved the integrity of the airways, the mechanisms involved and the links between these two mechanisms remained mysterious, which hindered the development of new therapies," explains Marc Chanson, a professor in the Department of Cell Physiology and Metabolism and the Geneva Center for Inflammation Research at the UNIGE Faculty of Medicine, who led this research.
Hydrating to restore tightness
The scientists first developed an in vitro model using human lung cells. This model, which was awarded the UNIGE 3R Prize in 2021 for reducing animal experimentation, reproduces airways epithelium of healthy and cystic fibrosis patients in a way that is both accurate and close to clinical reality. In collaboration with the team of Christian van Delden and Thilo Köhler from the Departments of Medicine and of Microbiology and Molecular Medicine at the UNIGE Faculty of Medicine, Marc Chanson and his team compared the response of epithelial cells invalidated for CFTR to bacterial infection, to which either hydrated, healthy mucus or physiological saline solution had been added.
"We observed a similar response in both cases: the presence of liquid, whatever its composition, restored the airways and protected them from infection," explains Juliette Simonin, post-doctoral fellow in Marc Chanson's laboratory and first author of the study. "Surface hydration is sufficient to tighten the junctions between cells and protects the epithelium integrity from bacterial colonization, even when CFTR is not functioning."
One treatment for all mutations?
A triple therapy pharmacologically targeting the CFTR protein has recently become available on the market. However, it only targets certain mutations of the CFTR gene and is only prescribed for a specific population of people with cystic fibrosis. More widely effective and safe treatments are still sorely lacking.
"Our results provide evidence that rehydration of the airway surface is beneficial. The challenge now is to find a simple way of doing this in all people with the disease, whatever the mutation involved," concludes Marc Chanson.
More information: Juliette L. Simonin et al, Surface Hydration Protects Cystic Fibrosis Airways from Infection by Restoring Junctional Networks, Cells (2022). DOI: 10.3390/cells11091587