This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
How the immune system becomes overactivated in old age

The lack of a key signaling molecule in certain immune cells can induce various age-related diseases in young mice, RIKEN researchers have shown. This finding eventually could help to develop new treatments for age-related diseases.
Aging isn't kind to our immune systems, causing over-activation of the system, so that inflammation occurs even when there are no pathogens in sight. Dubbed inflammaging, this chronic, low-grade inflammation damages tissue, making elderly people more susceptible to infection and a wide range of diseases, including cancer, type 2 diabetes and heart disease.
Inflammaging causes T cells—immune cells that recognize, respond to, and remember specific pathogens—to go into overdrive, producing inflammation-causing compounds such as cytokines and chemokines. This is known as the senescence state of T cells.
"People often have the impression that aging makes cells less active," says Takashi Saito of the RIKEN Center for Integrative Medical Sciences. "However, some aged immune cells enter an activated phase, and the resulting inflammaging can give rise to various age-dependent diseases."
A signaling molecule called receptor-interacting protein kinase 1 (RIPK1) controls cell death through two different paths, depending on which compounds it combines with. Recent studies had found that people with a RIPK1 deficiency are more susceptible to inflammatory disorders.
Now, Saito and co-workers have engineered mice that lack RIPK1 just in their T cells. They found that these mice developed inflammatory diseases at a young age and died much earlier than normal mice. Interestingly, the T cells lacking RIPK1 behaved similarly to T cells in aged mice. The study is published in the journal Science Advances.
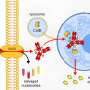
The team uncovered how this premature inflammaging occurs. When RIPK1 is not present, two compounds, caspase-8 and RIPK3, cause a cell-growth regulator known as mTORC1 to be excessively activated. This in turn promotes T cell senescence by inducing the expression of senescence-related genes, resulting in the production of various cytokines and chemokines.
This mechanism was unexpected. "Caspase-8 and RIPK3 are well known to induce cell death," says Saito. "But we've shown that they also help to activate mTORC1 and induce inflammaging in cellular senescence."
This discovery opens up the possibility of finding new ways to treat inflammaging. "We could target caspase-8 and RIPK3 to counteract inflammaging induced by T cell senescence," says Saito.
A further surprise was that when the team put the senescent T cells into normal mice, they reverted to normal, non-senescent T cells, indicating that environmental factors play an important role in regulating T cell senescence.
The team is now investigating what happens before activation of RIPK3 and caspase-8. They also want to explore what environmental factors allow senescent T cells to revert to normal T cells.
More information: Takayuki Imanishi et al, RIPK1 blocks T cell senescence mediated by RIPK3 and caspase-8, Science Advances (2023). DOI: 10.1126/sciadv.add6097