This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
Research finds rare disease shares mechanism with cystic fibrosis
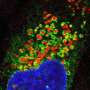
. In a patient afflicted with cystinosis, mutated cystine transporters are degraded by a cellular mechanism that degrades damaged proteins, called endoplasmic-reticulum-associated degradation. Consequently, the lysosome is unable to remove amino acid from the organelle, causing a build-up of cystine (center). Using a chemical chaperone also used to treat a form of cystic fibrosis, University of Michigan researchers were able to protect the mutated lysosome cystine transporter from degradation (right). The chemical chaperones aided in clearing cystine from the lysosome. Credit: U-M Ming Lab")
University of Michigan researchers have discovered that the same cellular mechanism involved in a form of cystic fibrosis is also implicated in a form of a rare disease called cystinosis.
The mechanism cleans up mutated proteins. In cystinosis, a genetic disease, this allows cystine crystals to build up in the cell. This disrupts the cell, and eventually disrupts tissues and ultimately organs, particularly the kidneys and the eyes.
The problem begins when the lysosome, an organelle within the cell, is unable to do its job. Often called the recycling center of the cell, the lysosome takes in cellular garbage, breaks it down into reusable cellular building blocks, then transports those materials back into the cell.
But when the protein that transports one of the recycled amino acids back into the cell mutates and fails, a cellular mechanism cleans up the faulty protein, allowing amino acid, or cystine, to build up in the lysosome.
The study findings are now published in the Journal of Clinical Investigation.
"If cystinosis not treated at an early age, some of the effects are irreversible and it could include impaired growth, kidney failure and neurological problems," said Varsha Venkatarangan, graduate student in the U-M Department of Molecular, Cellular and Developmental Biology and lead author of the study.
"Typically, the symptoms of the disease are treated rather than the root problem. So we have been wondering what could be the possible cellular mechanism of this disease."
Venkatarangan worked with fibroblasts, skin tissue cells derived from patients with the disease. Using these cells, she determined that this disease mechanism called endoplasmic-reticulum-associated degradation, or ERAD, degrades a mutated version of the lysosome cystine transporter.
ERAD is the same mechanism behind other diseases such as a major form of cystic fibrosis. And because this mechanism has been identified, the researchers were able to show that previously identified drug molecules were able to help the transporter protein remain stable.
"This drug molecule is a small chemical that can somehow assist the protein folding of the mutant and protect it from being degraded," said MCDB professor Ming Li. "This chemical chaperone helps it achieve the right formation so that it can be functional."
The lab, led by Li, became interested in the cystine transporting protein and wanted to understand how mutations in the transporter gene caused cystinosis. They painstakingly collected nearly 40 mutations that caused cystinosis and studied their protein stability. The researchers identified one striking mutation that had a very short half life compared to the non-mutated form of the protein.
"We were interested about the fact that this disease mutant was degrading so rapidly, what was the potential mechanism by which it was degrading, and the machinery involved," Venkatarangan said.

So Venkatarangan performed numerous genetic and chemical tests to inhibit different protein degradation machinery of the cell, narrowing down which cellular mechanism was involved in the disease. She determined that the mechanism was ERAD, a cellular pathway located in the cell's endoplasmic reticulum. The cell's endoplasmic reticulum is a network of tiny tubes and sheets that help ferry the cell's proteins to their destination. ERAD targets misfolded proteins and marks them for protein degradation.
Because this is a commonly known disease mechanism, involved in cystic fibrosis, drug molecules had already been identified to work around the ERAD mechanism and prolong the life of the protein. These drug molecules, Li said, are called chemical chaperones and help the protein achieve the right formation so that they can be functional.
"This small chemical chaperone can somehow magically—we don't know the exact mechanism—bind to our mutant and protect the protein from being degraded," Li said.
The researchers were able to stabilize the protein and determine that it was in the right location where it could function to ferry amino acids out of the lysosome, but they then needed to determine whether the transporter was still functional. Performing an assay in collaboration with UCSD, the researchers measured the cystine concentration in the lysosome. With a functional transporter, the lysosome would have lower amounts of amino acid.
"The reduction was pretty dramatic—nearly a 70% reduction of the accumulation level," Li said. "That directly brought the system level to almost normal. If we actually used this chemical chaperone to treat patients, it's possible that we could directly bring down the cystine accumulation in the lysosome."
People may wonder why a disease mechanism found in the endoplasmic reticulum might affect the function of lysosome membrane proteins, Li said. That's because lysosome membrane proteins are transported from the endoplasmic reticulum.
While the lysosome is being built, it needs a lot of trafficking proteins. These lysosomal proteins go through the endoplasmic reticulum and then the Golgi apparatus, which packages the proteins and sends them to the lysosome. During protein trafficking, they have to fold properly. Otherwise, they will be degraded prematurely by protein quality control systems such as ERAD.
These mutant proteins disappear early in the trafficking stage, the researchers discovered, after getting stuck and degraded in the endoplasmic reticulum.
In finding a new purpose for a drug already in use, the team also fulfilled a goal of a program at the National Institutes of Health that encourages finding new uses for already-approved drugs as this leads to faster therapies. However, the team emphasizes that the work is preliminary, done only in cultured patient cells, and the drug has not been tested to treat cystinosis in animals or humans.
"Again, this is preliminary work that was done in patient cells, so we don't really know the systemic effect or how well it works in actual patients," Venkatarangan said. "Our finding also emphasizes the importance of precision medicine, because most of the time, disease mutations are treated similarly under one umbrella of a particular disease.
"But the fact that individual mutations may actually have their own pathogenesis mechanism emphasizes that understanding the individual patient mechanism might help them with a better treatment strategy."
Co-authors of the paper include U-M MCDB's Weichao Zhang and Xi Yang. Jess Thoene of the Department of Pediatrics at the U-M Medical School and Si Houn Hahn of the Seattle Children's Hospital at University of Washington School of Medicine also contributed to the study.
More information: Varsha Venkatarangan et al, ER-associated degradation in cystinosis pathogenesis and the prospects of precision medicine, Journal of Clinical Investigation (2023). DOI: 10.1172/JCI169551