Researchers utilize genetically corrected stem cells to spark muscle regeneration
Researchers at the University of Minnesota's Lillehei Heart Institute have combined genetic repair with cellular reprogramming to generate stem cells capable of muscle regeneration in a mouse model for Duchenne Muscular Dystrophy (DMD).
The research, which provides proof-of-principle for the feasibility of combining induced pluripotent stem cell technology and genetic correction to treat muscular dystrophy, could present a major step forward in autologous cell-based therapies for DMD and similar conditions and should pave the way for testing the approach in reprogrammed human pluripotent cells from muscular dystrophy patients.
The research is published in Nature Communications.
To achieve a meaningful, effective muscular dystrophy therapy in the mouse model, University of Minnesota researchers combined three groundbreaking technologies.
First, researchers reprogrammed skin cells into "pluripotent" cells – cells capable of differentiation into any of the mature cell types within an organism. The researchers generated pluripotent cells from the skin of mice that carry mutations in the dystrophin and utrophin genes, causing the mice to develop a severe case of muscular dystrophy, much like the type seen in human DMD patients. This provided a platform that would mimic what would theoretically occur in human models.
The second technology employed is a genetic correction tool developed at the University of Minnesota: the Sleeping Beauty Transposon, a piece of DNA that can jump into the human genome, carrying useful genes with it. Lillehei Heart Institute researchers used Sleeping Beauty to deliver a gene called "micro-utrophin" to the pluripotent cells they were attempting to differentiate.
Much like dystrophin, human micro-utrophin can support muscle fiber strength and prevent muscle fiber injury throughout the body. But one key difference between the two is in how each is perceived by the immune system. Because dystrophin is absent in muscular dystrophy patients, its presence can prompt a devastating immune system response. But in those same patients, utrophin is active and functional, making it essentially "invisible" to the immune system. This invisibility allows the micro-utrophin to replace the dystrophin and progress the process of building and repairing muscle fiber within the body.
The third technology utilized is a method to produce skeletal muscle stem cells from pluripotent cells – a process developed in the laboratory of Rita Perlingeiro, Ph.D., the principal investigator of the latest study.
Perlingeiro's technology involves giving pluripotent cells a short pulse of a muscle stem cell protein called Pax3. The Pax3 protein pushes the pluripotent cells to become muscle stem cells, and allows them to be expanded exponentially in number. The Pax3-induced muscle stem cells were then transplanted back into the same strain of muscular dystrophy mice from which the pluripotent stem cells were originally derived.
Combined, the platforms created muscle-generating stem cells that would not be rejected by the body's immune system. According to Perlingeiro, the transplanted cells performed well in the dystrophic mice, generating functional muscle and responding to muscle fiber injury.
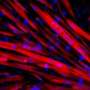
"We were pleased to find the newly formed myofibers expressed the markers of the correction, including utrophin," said Perlingeiro, a Lillehei endowed scholar within the Lillehei Heart Institute and an associate professor in the University of Minnesota Medical School. "However, a very important question following transplantation is if these corrected cells would self-renew, and produce new muscle stem cells in addition to the new muscle fibers."
By injuring the transplanted muscle and watching it repair itself, the researchers demonstrated that the cell transplants endowed the recipient mice with fully functional muscle stem cells.
This latest project from the U of M provides the proof-of-principle for the feasibility of combining induced pluripotent stem cell technology and genetic correction to treat muscular dystrophy.
"Utilizing corrected induced pluripotent stem cells to target this specific genetic disease proved effective in restoring function," said Antonio Filareto, Ph.D., a postdoctoral fellow in Perlingeiro's laboratory and the lead author on the study. "These are very exciting times for research on muscular dystrophy therapies."
These studies pave the way for testing this approach in reprogrammed human pluripotent cells from muscular dystrophy patients.
According to Perlingeiro, "Developing methods to genetically repair muscular dystrophy in human cells, and demonstrating efficacy of muscle derived from these cells are critical near-term milestones, both for the field and for our laboratory. Testing in animal models is essential to developing effective technologies, but we remained focused on bringing these technologies into use in human cells and setting the stage for trials in human patients."