Rare, lethal childhood disease tracked to protein
A team of international researchers led by Northwestern Medicine scientists has identified how a defective protein plays a central role in a rare, lethal childhood disease known as Giant Axonal Neuropathy, or GAN. The finding is reported in the May 2013 Journal of Clinical Investigation.
GAN is an extremely rare and untreatable genetic disorder that strikes the central and peripheral nervous systems of young children. Those affected show no symptoms at birth; typically around age three the first signs of muscle weakness appear and progress slowly but steadily. Children with GAN experience increasing difficulty walking and are often wheelchair-bound by age 10. Over time, they become dependent on feeding and breathing tubes. Only a few will survive into young adulthood.
In GAN patients, nerve cells are swollen with massive build-ups of structures called intermediate filaments, cytoskeletal components that give cells their shape and mechanical properties. Goldman's team found that gigaxonin, a protein encoded by the gene involved in GAN, regulates normal turnover of the protein building blocks that form a cell's intermediate filaments. Mutations in this gene result in the malfunctioning of gigaxonin, which leads to the abnormal build-up of intermediate filaments and eventually disrupts the normal functioning of nerve cells.
"This important new research pinpoints the mechanism that allows intermediate filaments to rapidly build up in GAN patients," says Robert Goldman, chair of the department of cell and molecular biology at Northwestern University Feinberg School of Medicine. Goldman has studied the structural proteins of cells for more than 30 years.
"This is a huge step forward for GAN research," said Lori Sames, co-founder and CEO of Hannah's Hope Fund, the leading GAN disease organization. "GAN is juvenile ALS, but even worse. Not only do motor neurons die out, so do the sensory neurons. To find a medicinal therapy, you really need to know what mechanism to target. And thanks to Dr. Goldman's work, now we do."
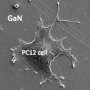
To identify gigaxonin's role, scientists used cells known as fibroblasts obtained from skin biopsies of children with GAN. The cells were then grown in lab cultures, and they also contained large abnormal aggregates of intermediate filaments. When scientists introduced healthy gigaxonin genes into both control and patient fibroblasts, the results were dramatic. The abnormal aggregates of intermediate filaments disappeared. However, the cytoskeleton's two other major systems, microtubules and actin filaments were not affected by this treatment.
The study's lead author, Northwestern University postdoctoral fellow Saleemulla Mahammad, stressed that this discovery may also have implications for more common types of neurodegenerative diseases that are also characterized by large accumulations of intermediate filament proteins, including Alzheimer's disease and Parkinson's disease.
"Our results suggest new pathways for disease intervention," he said. "Finding a chemical component that can clear the intermediate filament aggregations and restore the normal distribution of intermediate filaments in cells could one day lead to a therapeutic agent for many neurological disorders."
Mahammad and other members of the Goldman Laboratory collaborated with Puneet Opal, M.D., associate professor in the Ken and Ruth Davee department of neurology and cell and molecular biology, along with researchers in the laboratory of Pascale Bomont, at the INSERM neurological institute in Montpelier, France, and the laboratory of Jean-Pierre Julien at the Université Laval in Quebec, Canada.