May 22, 2014 report
The control of dendritic branching by mitochondria

(Medical Xpress)—A fundamental difference between neurons in real brains and those in artificial neural networks is the way the neurons in each are connected. In artificial nets, the synapses between neurons often have adjustable strengths, but the structure and scale of the input dendritic field generally counts for little. For real neurons, where a "connection" between cells is not just a synapse but rather a whole net unto itself, structure and scale are everything. The architect of this dendritic structure is neither a DNA code nor a spontaneous developmental physics that condenses order from a protein-lipid chaos. This structure is in fact the byproduct of competitive, yet cooperative mitochondria that administer that code to themselves and to their host to control its interaction with other similarly controlled hosts.
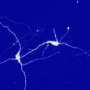
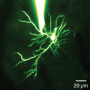
Reseachers from Osaku University have found that if mitochondria are depleted from developing dendrites in pyramidal cells, there is increased branching in the proximal region of the dendrites. In their paper last week in the Journal of Neuroscience, they also show that these dendrites grow longer. Since mitochondria distribute not just energy but also metabolites, proteins, and mRNAs throughout the cell, these results may be somewhat surprising. However depending on what manipulations have been done to alter the mitochondria, many things might be expected to happen to dendrites and the cell in general.
Previous reports have shown that eliminating mitochondria from dendrites of mitofusion-2 null Purkinje cells results in a reduction in dendrite number. Other studies in cultured hippocampal neurons found no alterations in branch patterns. The present study achieved depletion of mitochondria from dendrites by altering similar mitofusion (Mfn) proteins. In this case Mfn-1 was overexpressed, essentially initiating a massive mitochondrial fusion fest. This subsequently confines them to the soma. The authors were also able to achieve the same effects on dendrites by overexpressing TRAK2. This is a truncated form of a motor-adapter protein which would presumably interfere with transport along the cytoskeleton.

The authors suggest that a main difference with their new study may be that for the prior Purkinje cell experiments the dendrites were already developed, while here the mitochondria were perturbed at the outset. They offer three mechanisms—ATP generation, Ca2 buffering, and caspase activation—towards an explanation of their observed effects, which unfortunately, they tend to simplify as being either "positive or negative dendritic regulation."
For all we know, the behavior of mitochondria that have congealed into a synticium may be as different from the dispersed state as is the slime-mold from the individual free-roaming amoeboids, or from the radicalized hormone-crazed locust swarm from peaceable lone foragers. Imaging the fast contractions and depolarizations of individual mitochondria may now give more clues as to what they are doing then any given handfull of molecular indicators, but such studies have not yet been done in the fused state, if we may even call it that.
To return to the opening remarks regarding networks, we might reasonably assume that a fundamental principle used by real brains but ignored in the artificial, is that when one neuron wants to talk to another it uses an expansive net to do it. Similarly, a related principle would be that at increasing scales (neurite lengths and distances between cells) dendro-dendro connected neurons using graded potentials turn into directionally polarized axo-dendritic units that speak with spikes. If we accept that spikes are typically evoked from a proximal axon spike generator then this effectively yields more power to the centralized, and by default, synchronizing soma.
The role of mitochondria in controlling local structures like spines has been known for some time. An understanding of their control of the gross structure of dendritic or axonal trees is now only at the theoretical state. A better grasp of these effects can give insight not just into individual cells, but whole systems of the brain. A cetacean brain, for example, may have become numbed over evolutionary time to the slow olfactory or gustatory senses that might benefit from the more deliberating integrative properties of the long apical dendrites that would be afforded by a thick cortex. A thin but expansive cortex on the other hand, with small dendrites, fast processing times, and correspondingly more deep layer output cells may be ideal in echolocation.
Nearly every day brings new understanding of the effects of individual mitochondrial-associated proteins on neurons and their diseases. Earlier this week for example, we saw how the accumulation in mitochondria of Huntingtin, a protein critical in Huntington's disease, leads to problems for the cell. As these novel mechanisms are clothed in ever greater molecular detail, it is important to keep the big picture in mind at the same time to wring all that we might from these experiments.
More information: Evidence That Dendritic Mitochondria Negatively Regulate Dendritic Branching in Pyramidal Neurons in the Neocortex, The Journal of Neuroscience, 14 May 2014, 34(20): 6938-6951; DOI: 10.1523/JNEUROSCI.5095-13.2014 , www.jneurosci.org/content/34/20/6938.abstract
Abstract
The precise branching patterns of dendritic arbors have a profound impact on information processing in individual neurons and the brain. These patterns are established by positive and negative regulation of the dendritic branching. Although the mechanisms for positive regulation have been extensively investigated, little is known about those for negative regulation. Here, we present evidence that mitochondria located in developing dendrites are involved in the negative regulation of dendritic branching. We visualized mitochondria in pyramidal neurons of the mouse neocortex during dendritic morphogenesis using in utero electroporation of a mitochondria-targeted fluorescent construct. We altered the mitochondrial distribution in vivo by overexpressing Mfn1, a mitochondrial shaping protein, or the Miro-binding domain of TRAK2 (TRAK2-MBD), a truncated form of a motor-adaptor protein. We found that dendritic mitochondria were preferentially targeted to the proximal portion of dendrites only during dendritic morphogenesis. Overexpression of Mfn1 or TRAK2-MBD depleted mitochondria from the dendrites, an effect that was accompanied by increased branching of the proximal portion of the dendrites. This dendritic abnormality cannot be accounted for by changes in the distribution of membrane trafficking organelles since the overexpression of Mfn1 did not alter the distributions of the endoplasmic reticulum, Golgi, or endosomes. Additionally, neither did these constructs impair neuronal viability or mitochondrial function. Therefore, our results suggest that dendritic mitochondria play a critical role in the establishment of the precise branching pattern of dendritic arbors by negatively affecting dendritic branching.
© 2014 Phys.org