Small molecule may play big role in Alzheimer's disease

Alzheimer's disease is one of the most dreaded and debilitating illnesses one can develop. Currently, the disease afflicts 6.5 million Americans and the Alzheimer's Association projects it to increase to between 11 and 16 million, or 1 in 85 people, by 2050.
Cell death in the brain causes one to grow forgetful, confused and, eventually, catatonic. Recently approved drugs provide mild relief for symptoms but there is no consensus on the underlying mechanism of the disease.
"We don't know what the problem is in terms of toxicity," said Joan-Emma Shea, professor of chemistry and biochemistry at the University of California, Santa Barbara (UCSB). "This makes the disease difficult to cure."
Accumulations of amyloid plaques have long been associated with the disease and were presumed to be its cause. These long knotty fibrils, formed from misfolded protein fragments, are almost always found in the brains of diseased patients. Because of their ubiquity, amyloid fibrils were considered a potential source of the toxicity that causes cell death in the brain. However, the quantity of fibrils does not correspond with the degree of dementia and other symptoms.
New findings support a hypothesis that fibrils are a by-product of the disease rather than the toxic agent itself. This paradigm shift changes the focus of inquiry to smaller, intermediate molecules that form and dissipate quickly. These molecules are difficult to perceive in brain tissue.
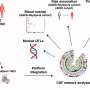
Shea's group uses computer simulations to understand the formation of toxic entities in the brain. Since 2007, Shea has run thousands of simulations of amyloid peptides using the Ranger supercomputer at the Texas Advanced Computing Center (TACC) to better understand the structure, formation and behavior of amyloid accumulations.
"We can identify the important structures or conformations that are adopted by these peptides at a resolution that exceeds what can be done experimentally," she explained. "This helps us understand what structures lead to a self-assembly."
For decades, it was believed that fibrils were a toxic species, but increasingly researchers are looking at small, soluble precursor forms of the fibrils, known as oligomers. "These are difficult to detect experimentally because they tend to be transient species," Shea said "There's no consensus on how big they are. There are still a lot of debates."
Shea and Michael Bowers, professor of chemistry and biochemistry at UCSB and Shea's experimental collaborator, believe the transient oligomers may be responsible for the onset of the disease through interactions with the cell membrane.
"These oligomers may be toxic by inserting themselves into membranes and causing a damage to the membrane," she said. "The membrane is critical for the cell viability."
In 2007, Shea and Bowers received a grant from the National Institutes of Health to investigate this theory. Together, they have spent the last five years looking at small peptide-based inhibitors that would prevent these oligomers from forming.
"If you can prevent the oligomers from forming, you can limit toxicity," Shea said.
In a recent paper currently in press in Biophysical Journal, Shea and postdoctoral researcher Luca Larini studied the conformations adopted by small oligomers of peptide amyloids encountered within the cell. They found that hairpin-shaped forms of the peptide initiated the aggregation of oligomers that ultimately led to the formation of a fibril. Like an old slapstick routine where one person trips, another trips over them, and eventually a pile forms, the misfolded proteins in the brain cells of those with Alzheimer's recruit other misfolded proteins and eventually grow into a large mass.
Shea's simulations have not only helped uncover the possible role of oligomers in the onset of Alzheimer's, but they are aiding in research that is trying to stop oligomer formation in the first place. A paper in the November 2011 edition of Biochemistry, co-authored with the Bowers group, described how a class of small molecules known as c-terminal inhibitors was able to stop the formation of oligomers, possibly halting disease progression before it is too late.
"Dr. Shea's simulations put a molecular face on the cross sections and oligomer distributions that we experimentally measure," said Bowers. " Of significant importance is the simulation of the ABeta42 monomer structure that very nicely correlated with our experiments. Also of importance are calculations on the sites and mechanism of attachment of potential therapeutic agents that we are testing as ABeta aggregation inhibitors."
Simulations on Ranger helped researchers identify where the inhibitors bind and led to new ideas about how inhibition can be improved.
"Dr. Shea is clearly at the top of the large cohort of simulators in her age group," Bowers said.
Through a related investigation, Shea and postdoctoral researcher Chun Wu solved the long-standing mystery of why Thioflavin T, a dye commonly used in brain imaging, is able to bind to amyloid proteins. Her molecular dynamics simulations identified the specific hydrophobic motif in the peptide to which the dye binds. This pinpoint conclusion now allows chemists and neurological experimentalists to create designer forms of the dye that can be used to improve their diagnostic ability. These results were reported in the Biophysical Journal in March 2011.
"Now that we've established where these molecules bind, we can start tweaking the molecule to try to make binders that have a greater affinity for the fibril. That could be something that would be beneficial for medicine as a better imaging agent," she said.
Shea's simulations of peptide interactions, dyes binding to fibrils, and inhibitors stopping the accumulation of amyloids provide great insights to scientists. The projects required more than 13 million hours of compute time on TACC's Ranger and Lonestar supercomputers since 2009.
"The number of atoms is huge—we need a lot of computational resources to simulate them," Shea said. "Nothing that we're doing here is something that we could do on our home clusters. The scale of it is intractable."
Ranger is one of the top 50 most powerful supercomputers in the world, Funded by the National Science Foundation and deployed in 2008, Ranger helps scientists around the country make discoveries by offering free compute time to academic researchers. The system is part of the Extreme Science and Engineering Discovery Environment (XSEDE), the NSF-funded effort to provide cyberinfrastructure and computing power to the nation's scientists.
In February, Ranger will be decommissioned to make way for Stampede, a new supercomputer 20 times more powerful. Such a system will be required to answer further important questions about Alzheimer's disease.
"With growing computational resources and capabilities, we'll be able to look at how these proteins interact with membranes," Shea said. "We're far away from simulating a whole cell, but we can start incorporating additional elements that may turn out to be important."