For mitochondria, bigger may not be better
Goldilocks was on to something when she preferred everything "just right." Harvard Medical School researchers have found that when it comes to the length of mitochondria, the power-producing organelles, applying the fairy tale's mantra is crucial to the health of a cell. More specifically, abnormalities in mitochondrial length promote the development of neurodegenerative diseases such as Alzheimer's.
"There had been a fair amount of interest in mitochondria in Alzheimer's and tau-related diseases, but causality was unknown," said Brian DuBoff, first author of the study and a post-doctoral research fellow at Massachusetts General Hospital.
"Ultimately, a deeper understanding of the relationship between mitochondrial function and Alzheimer's may guide us to develop more targeted therapies in the future," said Mel Feany, HMS professor of pathology at Brigham and Women's Hospital and senior author of the paper.
The findings will be published online in the August 23 issue of Neuron.
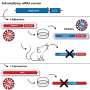
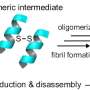
Tau-related diseases are caused when tau, a protein most commonly found in neurons, malfunctions. Tau binds to microtubules in cells, a process known as stabilization. This binding is necessary so the microtubules can help maintain cell structure and aid in intracellular processes such as transporting molecules. When tau is defective, most often due to changes introduced during protein synthesis, it can accumulate in neurofibrillary tangles, one of the primary markers of Alzheimer's.
In this particular study, conducted in fruit flies with defective tau protein, DuBoff found that the mitochondria in the brain cells of these flies were elongated compared with the mitochondria in flies with normal tau. The elongation, he observed, adversely affected mitochondrial function.
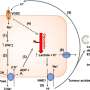
"Normally, one mitochondrion will split into two, two mitochondria will join into one, and that's a critical process for the health and stability of the mitochondria," said DuBoff. "This mitochondrial dynamic happens continuously in almost all cells. Interruption of this process leads to cell death, and loss of nerve cells in the brain results in loss of function—memory loss and difficulty in comprehension and coordination." The presence of defective tau, then, interrupts the functioning of mitochondria and contributes to neurodegeneration.
To further observe how mitochondrial dynamics were affected by the presence of defective tau, the researchers modified two sets of genes in human-tau-expressing flies, one that controls how mitochondria divide and another that guides how they come together. When the expression of the gene that causes mitochondrial lengthening, or fusion, was increased, the level of neurodegeneration in the flies increased and the flies were sicker. Conversely, when the expression of the gene that causes mitochondrial division, or fission, was increased, the defect reversed and the flies' condition improved.
The study also showed that, in addition to tau, two other key proteins influenced the neurodegenerative process: DRP1, which helps in the fission of mitochondria, and actin, which is essential to maintaining cell structure and movement. A previous study in Feany's lab had shown that the presence of defective tau hampers the activity of actin. With this knowledge, the researchers were able to piece together the relationship among the three proteins. DRP1 and actin are interdependent: the regulatory state of actin is essential for DRP1 and mitochondria to come together, thus preserving mitochondrial dynamics. But the presence of defective tau harms this relationship, rendering DRP1 incapable of maintaining mitochondrial dynamics, which ultimately leads to neurodegeneration.
"We have a good idea now of where the process starts. We know it ends with neurodegeneration, and with this study, we know some milestones along the way," said Feany. "But we still have to fill in the gaps and learn more about DRP1 and its role in this process."
"Many studies begin by looking at a normal biological process and then finding ways it goes wrong," said DuBoff. "We did the opposite. We started with the disease model, identified this phenomenon of DRP1 and mitochondrial dysfunction, and then followed it back to the basic biological regulation of this process."