Scientists discover that DNA damage occurs as part of normal brain activity
Scientists at the Gladstone Institutes have discovered that a certain type of DNA damage long thought to be particularly detrimental to brain cells can actually be part of a regular, non-harmful process. The team further found that disruptions to this process occur in mouse models of Alzheimer's disease—and identified two therapeutic strategies that reduce these disruptions.
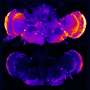
Scientists have long known that DNA damage occurs in every cell, accumulating as we age. But a particular type of DNA damage, known as a double-strand break, or DSB, has long been considered a major force behind age-related illnesses such as Alzheimer's. Today, researchers in the laboratory of Gladstone Senior Investigator Lennart Mucke, MD, report in Nature Neuroscience that DSBs in neuronal cells in the brain can also be part of normal brain functions such as learning—as long as the DSBs are tightly controlled and repaired in good time. Further, the accumulation of the amyloid-beta protein in the brain—widely thought to be a major cause of Alzheimer's disease—increases the number of neurons with DSBs and delays their repair.
"It is both novel and intriguing team's finding that the accumulation and repair of DSBs may be part of normal learning," said Fred H. Gage, PhD, of the Salk Institute who was not involved in this study. "Their discovery that the Alzheimer's-like mice exhibited higher baseline DSBs, which weren't repaired, increases these findings' relevance and provides new understanding of this deadly disease's underlying mechanisms."
In laboratory experiments, two groups of mice explored a new environment filled with unfamiliar sights, smells and textures. One group was genetically modified to simulate key aspects of Alzheimer's, and the other was a healthy, control group. As the mice explored, their neurons became stimulated as they processed new information. After two hours, the mice were returned to their familiar, home environment.
The investigators then examined the neurons of the mice for markers of DSBs. The control group showed an increase in DSBs right after they explored the new environment—but after being returned to their home environment, DSB levels dropped.
"We were initially surprised to find neuronal DSBs in the brains of healthy mice," said Elsa Suberbielle, DVM, PhD, Gladstone postdoctoral fellow and the paper's lead author. "But the close link between neuronal stimulation and DSBs, and the finding that these DSBs were repaired after the mice returned to their home environment, suggest that DSBs are an integral part of normal brain activity. We think that this damage-and-repair pattern might help the animals learn by facilitating rapid changes in the conversion of neuronal DNA into proteins that are involved in forming memories."
The group of mice modified to simulate Alzheimer's had higher DSB levels at the start—levels that rose even higher during neuronal stimulation. In addition, the team noticed a substantial delay in the DNA-repair process.
To counteract the accumulation of DSBs, the team first used a therapeutic approach built on two recent studies—one of which was led by Dr. Mucke and his team—that showed the widely used anti-epileptic drug levetiracetam could improve neuronal communication and memory in both mouse models of Alzheimer's and in humans in the disease's earliest stages. The mice they treated with the FDA-approved drug had fewer DSBs. In their second strategy, they genetically modified mice to lack the brain protein called tau—another protein implicated in Alzheimer's. This manipulation, which they had previously found to prevent abnormal brain activity, also prevented the excessive accumulation of DSBs.
The team's findings suggest that restoring proper neuronal communication is important for staving off the effects of Alzheimer's—perhaps by maintaining the delicate balance between DNA damage and repair.
"Currently, we have no effective treatments to slow, prevent or halt Alzheimer's, from which more than 5 million people suffer in the United States alone," said Dr. Mucke, who directs neurological research at Gladstone and is a professor of neuroscience and neurology at the University of California, San Francisco, with which Gladstone is affiliated. "The need to decipher the causes of Alzheimer's and to find better therapeutic solutions has never been more important—or urgent. Our results suggest that readily available drugs could help protect neurons against some of the damages inflicted by this illness. In the future, we will further explore these therapeutic strategies. We also hope to gain a deeper understanding of the role that DSBs play in learning and memory—and in the disruption of these important brain functions by Alzheimer's disease."