Study offers new approach for spinal muscular atrophy
Spinal muscular atrophy is a debilitating neuromuscular disease that in its most severe form is the leading genetic cause of infant death. By experimenting with an ALS drug in two very different animal models, researchers at Brown University and Boston Children's Hospital have identified a new potential mechanism for developing an SMA treatment.
There is no specific drug to treat spinal muscular atrophy (SMA), a family of motor neuron diseases that in its most severe form is the leading genetic cause of infant death in the United States and affects one in 6,000 people overall. But a new multispecies study involving a drug that treats amyotrophic lateral sclerosis (ALS) has pinpointed a mechanism of SMA that drug developers might be able to exploit for a new therapy.
The research, published in the Journal of Neuroscience, reports that the drug Riluzole advanced neural cell development in a mammalian model of SMA and restored neuromuscular function and mobility in a Caenorhabditis elegans worm model of the disease.
Riluzole has already been tested as a therapy in a very small study of severely affected SMA patients. It failed to help. Nevertheless, what makes the new research encouraging, said Anne Hart, professor of neuroscience at Brown and senior author on the paper, is that the study traces the beneficial action of Riluzole to specific "SK2" potassium channels in worm neurons. Humans have these channels too, and if they can be more precisely targeted by a new drug, she said, that could make a more meaningful difference, at least for some patients.
"We're not suggesting based on this that SMA patients should ask their doctors for Riluzole," said Hart, who is affiliated with the Brown Institute for Brain Science, "but we are suggesting that this pathway would be useful for therapeutic development."
How Riluzole works
Because SMA has a lot in common with ALS, Hart thought Riluzole might still be worth studying in the context of SMA. To do so, she partnered with fellow researchers at Boston Children's Hospital. They worked in mouse neuronal cells while her team at Brown worked in the worms.
For each system, the researchers created SMA models in different ways by disabling the gene that produces the survival motor neuron (SMN) protein. Depletion of that protein causes SMA in people too.
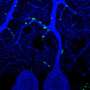
In the mammalian neuronal cells, the Children's Hospital researchers found that Riluzole promoted the growth of axons that was lacking in the SMN-depleted cells. However, Riluzole did this not by increasing SMN levels. Instead, the researchers found evidence that drug treatment matured the neurons more quickly in normal cells.
Most attempts to treat SMA have relied on trying to maintain or restore higher levels of SMN, Hart noted. But Riluzole, or a future drug, may instead be able to work by accelerating cell maturity.
In the worms meanwhile, the Brown researchers found that Riluzole restored two important neuromuscular behaviors of SMA worms: the pumping action that allows the worms to move food through their digestive tracts and the body bending that they perform when swimming.
To learn how Riluzole had this effect, they performed further experiments testing various potassium channels, including SK2, that Riluzole is known to act upon. Losing these channels didn't cause more problems in animals with less SMN protein, but losing the SK2 potassium channels in particular made neuromuscular function worse. Without the SK2 channels, the drug Riluzole didn't improve function.
"This told us that Riluzole improves motorneuron function by acting through SK2 channels, which we did not know before," said lead author Maria Dimitriadi, a postdoctoral researcher in Hart's group. "This is important because understanding how SK2 channels affect motorneuron disease may eventually lead to a treatment helping both ALS and SMA patients."