Jammed molecular motors may play a role in the development of ALS
Slowdowns in the transport and delivery of nutrients, proteins and signaling molecules within nerve cells may contribute to the development of the neurodegenerative disorder ALS, according to researchers at the University of Illinois at Chicago College of Medicine.
The researchers showed how a genetic mutation often associated with inherited ALS caused delays in the transport of these important molecules along the long axons of neurons.
Their findings were published in the online journal PLOS ONE on June 12.
Motor neurons are among the longest cells in the human body—some may extend half a person's height, as much as three feet. This poses a problem if all the cellular building blocks are made at one end of the cell, where the nucleus sits, but are needed at the other end of the cell.
Neurons have the molecular equivalents of highways and delivery trucks—nerve fibers and motor proteins—that run along their long axons, ferrying material back and forth. But when shipping is held up, and products aren't getting to where they are needed, the cell can't function optimally. These transport problems can cause neurons to lose contact with other neurons and muscles.
"If the transport process is delayed or slowed, the terminal end of the cell can run out of materials it needs, and can lose its synaptic connection with its neighboring neurons," says Gerardo Morfini, UIC assistant professor of anatomy and cell biology and the co-principal investigator on the study. "Without the connections, the cells die."
"Cell death is the final stage in a long disease process in ALS," said Scott Brady, UIC professor and head of anatomy and cell biology and co-principal investigator. "We wanted to understand the pathological process in neurons leading up to cell death."
Neuroscientists know that mutations in a protein called SOD1 account for many of the 10 percent of ALS cases that are inherited. Ninety percent of ALS cases have no known cause and are termed sporadic.
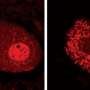
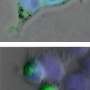
Brady and colleagues had previously shown, using high-resolution video microscopy of squid axons, that a mutant variant of the protein significantly slowed down the transport of material from one end of the cell to the other.
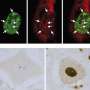
In the new study, the researchers looked at how the mutated form of SOD1 caused the slowdown in cellular transport. They found that the mutated protein activated molecules called p38 kinases, which in turn modified a major motor protein involved in moving cargo along the nerve axons. These modified motor proteins moved poorly compared to controls that were exposed to unmutated SOD1.
They also showed that transport in in genetically altered mice was also slowed by mutant SOD1, through the same mechanism.
"The pathways between SOD1 and the p38 kinases could provide interesting targets for therapeutic intervention in treating ALS, both for some of the genetic forms and the spontaneous forms, where malfunctioning SOD1 is also a contributing factor," said Brady.