Tuberculosis and Parkinson's disease linked by unique protein
A protein at the center of Parkinson's disease research now also has been found to play a key role in causing the destruction of bacteria that cause tuberculosis, according to scientists led by UC San Francisco microbiologist and tuberculosis expert Jeffery Cox, PhD.
The protein, named Parkin, already is the focus of intense investigation in Parkinson's disease, in which its malfunction is associated with a loss of nerve cells. Cox and colleagues now report that Parkin also acts on tuberculosis, triggering destruction of the bacteria by immune cells known as macrophages. Results appear online today (September 4, 2013) in the journal Nature.
The finding suggests that disease-fighting strategies already under investigation in pre-clinical studies for Parkinson's disease might also prove useful in fighting tuberculosis, according to Cox. Cox is investigating ways to ramp up Parkin activity in mice infected with tuberculosis using a strategy similar to one being explored by his UCSF colleague Kevan Shokat, PhD, as a way to ward off neurodegeneration in Parkinson's disease.
Globally, tuberculosis kills 1.4 million people each year, spreading from person to person through the air. Parkinson's disease, the most common neurodegenerative movement disorder, also affects millions of mostly elderly people worldwide.
Cox homed in on the enzyme Parkin as a common element in Parkinson's and tuberculosis through his investigations of how macrophages engulf and destroy bacteria. In a sense the macrophage—which translates from Greek as "big eater"—gobbles down foreign bacteria, through a process scientists call xenophagy.
Mycobacterium tuberculosis, along with a few other types of bacteria, including Salmonella and leprosy-causing Mycobacterium leprae, are different from other kinds of bacteria in that, like viruses, they need to get inside cells to mount a successful infection.
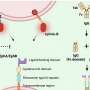
The battle between macrophage and mycobacterium can be especially intense. M. tuberculosis invades the macrophage, but then becomes engulfed in a sac within the macrophage that is pinched off from the cell's outer membrane. The bacteria often escape this intracellular jail by secreting a protein that degrades the sac, only to be targeted yet again by molecular chains made from a protein called ubiquitin. Previously, Cox discovered molecules that escort these chained mycobacteria to more secure confinement within compartments inside cells called lysosomes, where the bacteria are destroyed.
The cells of non-bacterial organisms ranging in complexity from baker's yeast to humans also use a similar mechanism—called autophagy—to dispose of their own unneeded molecules or worn out cellular components. Among the most abundant and crucial of these components are the cell's mitochondria, metabolic powerhouses that convert food molecules into a source of energy that the cell can readily use to carry out its everyday housekeeping chores, as well as its more specialized functions.
Like other cellular components, mitochondria can wear out and malfunction, and often require replacement. The process through which mitochondria are disposed of, called mitophagy, depends on Parkin.
Cox became curious about the enzyme when he learned that specific, naturally occurring variations in the Parkin gene, called polymorphisms, are associated with increased susceptibility to tuberculosis infection.
"Because of the commonalities between mitophagy and the xenophagy of intracellular mycobacteria, as well as the links between Parkin gene polymorphisms and increased susceptibility to bacterial infection in humans, we speculated that Parkin may also be recruited to M. tuberculosis and target it for xenophagy," Cox said.
In both mouse and human macrophages infected with M. tuberculosis in the lab, Parkin played a key role in fighting the bacteria, Cox and colleagues found. In addition, genetically engineered mice lacking Parkin died when infected with M. tuberculosis, while mice with normal Parkin survived infection.
The involvement of Parkin in targeting both damaged mitochondria and infectious mycobacteria arose long ago in evolution, Cox argues. As part of the Nature study, the research team found that Parkin-deficient mice and flies – creatures quite distant from humans in evolutionary time – also are more sensitive than normal mice and flies to intracellular bacterial infections.
Looking back more than 1 billion years, Cox noted that mitochondria evolved from bacteria that were taken up by cells in a symbiotic relationship.
In the same way that the immune system recognizes infectious bacteria as foreign, Cox said, "The evolutionary origin of mitochondria from bacteria suggests that perhaps mitochondrial dysfunction triggers the recognition of a mitochondrian as non-self."
Having now demonstrated the importance of Parkin in fighting mycobacterial infection, Cox has begun working with Shokat to find a way to boost Parkin activity against cell-invading bacteria. "We are exploring the possibility that small-molecule drugs could be developed to activate Parkin to better fight tuberculosis infection," Cox said.