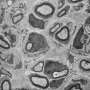
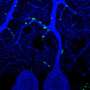
Schwann cells 'dine in' to clear myelin from injured nerves
. The myelin sheath breaks up into fragments in the cytoplasm that are transported by autophagosomes to lysosomes for degradation. Credit: Gomez-Sanchez et al., 2015")
Researchers reveal how cells in the peripheral nervous system (PNS) degrade myelin after nerve injury, a process that fails to occur in the central nervous system (CNS). The study appears in The Journal of Cell Biology.
Unlike the CNS, the PNS is capable of significant repair. One reason for this is that Schwann cells, which supply the protective sheath of myelin that covers axons in the PNS, are able to help clear away damaged myelin and guide regeneration after nerve injury. In contrast, the myelin-forming cells in the brain and spinal cord, oligodendrocytes, lack these properties, which helps explains why the CNS has such a limited ability to fix damaged nerves.
The cellular mechanism that enables Schwann cells to break down their own myelin is unclear. This process is key to understanding and treating diseases resulting in myelin damage (demyelination), which can be caused by inherited mutations, illness, age, or injuries.
Researchers previously thought that demyelination by Schwann cells might occur through phagocytosis, whereby the cells gobble up extracellular myelin. But a new study, led by Ashwin Woodhoo (CIC bioGUNE-CIBERehd & Ikerbasque, Spain) and Kristján Jessen (University College London), shows otherwise.
Although Schwann cells and immune cells may employ phagocytosis later in the cleanup process, the new findings indicate that in the first 5-7 days after injury, when 40-50% of the myelin is cleared, Schwann cells digest myelin internally. This cleanup appears to take place through autophagy, with myelin debris inside of Schwann cells being carried to lysosomes in a process the authors term "myelinophagy."
Using mouse sciatic nerves, which are part of the PNS, the researchers found that expression of several autophagy-related genes was induced after injury. Four to seven days after injury, they also observed an abundance of autophagosomes—which deliver cargo to lysosomes for degradation—compared with uninjured nerves, and these autophagosomes contained myelin debris. Inhibiting autophagy led to a notable reduction in myelin breakdown.
The authors also studied mutant mice lacking autophagy function. Although the mutants survived normally and their peripheral nerves were indistinguishable from control mice, they displayed a significant reduction in myelin breakdown after injury compared with the control group. Using a mouse model of Charcot-Marie-Tooth disease, the most common hereditary demyelinating disease, they observed elevated levels of autophagy compared with healthy nerves.
Because myelin breakdown in the CNS is ineffective, the researchers also examined an injured optic nerve. Key indicators of autophagy were not elevated in the optic nerve in contrast to the sciatic, suggesting that the failure of the CNS to clear myelin is associated with reduced activation of autophagy.
"Overall, the findings indicate that autophagy is a mechanism for myelin breakdown in Schwann cells and could provide new targets for manipulating demyelination in injury and disease," says Jessen. "The results also shed new light on the remarkable absence of myelin breakdown in the CNS."
"Further studies are needed to determine if modulating myelinophagy can prevent myelin breakdown in demyelinating diseases or improve nerve repair after PNS and CNS injury," says Woodhoo.
More information: Gomez-Sanchez, J.A., et al. 2015. J. Cell Biol. DOI: 10.1083/jcb.201503019