Study focuses on 'speed bumps' protecting against Alzheimer's disease

Dedicated to the study of Alzheimer's disease, University of Virginia biology professor George Bloom's lab has spent the last decade working to expand the scientific community's still-primitive understanding of the disease's underlying biology. A recently published study from Bloom's lab is helping to solve one of the devastating brain disorder's underlying mysteries, explaining how certain proteins convert previously healthy nerve cells into neurons afflicted with Alzheimer's.
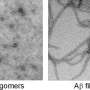
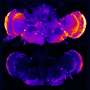
Other labs have previously shown how aggregations of a neuronal protein called "tau," amassing in the slender, threadlike form of filaments, constitute the "neurofibrillary" tangles of brains afflicted with Alzheimer's disease.
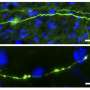
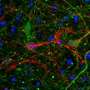
The new UVA study revealed, however, that much smaller aggregates of tau outside brain cells, known as "extracellular tau oligomers," cause the tau inside neurons not only to aggregate, but to spread from its normal location in axons (the wire-like extensions of neurons), into the branched, tree-like parts of a neuron called dendrites.
The significance of this finding – described in a Journal of Alzheimer's Disease article by lead researcher Eric Swanson, Bloom and four other UVA-affiliated authors – is that other labs showed previously that excess dendritic tau poisons the synapses, or connections, among neurons that control memory and cognition. The work of Swanson and his colleagues revealed that extracellular tau oligomers, which are known to accumulate in the brains of Alzheimer's disease patients, cause intracellular tau to invade dendrites, and by extension, compromise synapses.
Swanson's finding reinforces the idea that the detection of tau oligomers may prove to be a key to diagnosing Alzheimer's disease at a much earlier stage than currently possible.
"No disease-modifying therapies currently exist for treating Alzheimer's, and our research provides further evidence for extracellular tau oligomers as a promising therapeutic target," said Swanson, who completed his Ph.D. at UVA earlier this spring. "The detection of these pathogenic species early in the disease's progression could also allow for therapeutic interventions before irreversible neuron death occurs, when it may be too late for treatment."
Bloom said their study found that oligomers are much better than filaments at causing the tau inside neurons to aggregate.
"This is the first study that we are aware of that addresses the consequences to the neuron related to the tau aggregating inside the neuron," Bloom said.
In addition to Swanson and Bloom, who also directs the College and Graduate School of Arts & Sciences' undergraduate neuroscience program, the six credited authors of the June 5 article include two UVA undergraduate researchers.
According to Bloom, Alzheimer's disease – at a projected cost of nearly $260 billion for the current year – has been more expensive to American society than cancer and heart disease in each year since 2013. Unless new, disease-modifying drugs can be developed soon, Bloom said, the financial burden imposed by Alzheimer's will continue to rise steadily and could increase as much as threefold by the middle of the century.
The tau oligomers identified in Swanson's study, however, represent potential new drug targets to combat the disease.
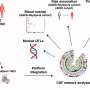
The article discusses another important discovery by Swanson, related to tau's role as "speed bumps" along microtubules, the long, intracellular fibers that serve as highways for transporting subcellular cargoes from place to place within cells. The tau inside axons is normally stuck to these microtubules, and previous studies have shown how tau serves as speed bumps, regulating how fast and far cargoes can move along microtubules.
According to Swanson's findings, when this axonal tau within the neuron is exposed to extracellular tau oligomers, the transport rate speeds up, and subcellular cargo moves along microtubules for distances longer than normal. This implies that once aggregated, the axonal tau falls off of the microtubules, causing the tau speed bumps to disappear.
"That's fully consistent with the idea that as tau is aggregating; it's falling off the microtubules," Bloom said. "Think of it as a railroad track with side tracks running off to deliver goods. So, if you were to get rid of the tracks leading off the main track, then all the cargo would be biased toward the end of the track, or the end of the axon. If that happens, then that axon might not receive all of the replacement parts that it needs.
"We haven't proven anything beyond that, but we suspect that disruption of axonal transport will gradually lead to a breakdown of what is called neuronal homeostasis. In other words, for a neuron to be in a steady state of high functional capacity, it's got to have just the right kind of axonal transport. If things are moving too fast in axons and for distances that are in excess of normal, then the delivery of that cargo will be biased toward the end of the axon and that will, we suspect, gradually cause a breakdown of the normal healthy axon."
Since the health of the neuron depends on axonal transport occurring normally, Swanson's results suggest that axonal transport, and the overall health of a neuron, can be poisoned by extracellular tau oligomers. Moving forward, this finding highlights the attractiveness of tau oligomers as therapeutic targets for Alzheimer's disease, Bloom said.
The Journal of Alzheimer's Disease published the article, "Extracellular Tau Oligomers Induce Invasion of Endogenous Tau into the Somatodendritic Compartment and Axonal Transport Dysfunction," in its June 5 edition (vol. 58, No. 3, pp. 803-820).
More information: Eric Swanson et al. Extracellular Tau Oligomers Induce Invasion of Endogenous Tau into the Somatodendritic Compartment and Axonal Transport Dysfunction, Journal of Alzheimer's Disease (2017). DOI: 10.3233/JAD-170168