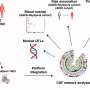
Alzheimer's gene poses both risk and benefits
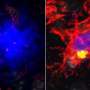
 in the brains of mice with the gene TREM2 (left) than in those without the gene (right). A new study shows that having a working copy of the gene TREM2 can reduce risk of Alzheimer's disease under certain conditions but worsen disease in others. Credit: David Holtzman Lab")
Scientists drilling down to the molecular roots of Alzheimer's disease have encountered a good news/bad news scenario. A major player is a gene called TREM2, mutations of which can substantially raise a person's risk of the disease. The bad news is that in the early stages of the disease, high-risk TREM2 variants can hobble the immune system's ability to protect the brain from amyloid beta, a key protein associated with Alzheimer's.
The good news, however, according to researchers at Washington University School of Medicine in St. Louis, is that later in the disease, when the brain is dotted with toxic tangles of another Alzheimer's protein known as tau, the absence of TREM2 protein seems to protect the brain from damage. Mice without TREM2 suffer much less brain damage than those with it.
The findings potentially make targeting the TREM2 protein as a means of preventing or treating the devastating neurodegenerative disease a little more complicated, and suggest that doctors may want to activate TREM2 early in the disease and tamp it down later.
"People in the Alzheimer's field have already been trying to develop ways to target TREM2," said senior author David Holtzman, MD, the Andrew B. and Gretchen P. Jones Professor and head of the Department of Neurology. "Now that we have this data, the question is, 'What does one really want to do? Stimulate it or inhibit it?'"
The study is published online the week of Oct. 9 in Proceedings of the National Academy of Sciences.
Amyloid beta plaques start forming in the brains of Alzheimer's patients years before the characteristic symptoms of memory loss and confusion appear. The plaques themselves appear to do minimal damage - many older people remain mentally sharp despite plentiful plaques - but their presence raises the risk of developing tau tangles, the real engine of destruction. The brain starts to die specifically in the areas where tau tangles are found.
In the brain, the protein TREM2 is found only on immune cells known as microglia. Holtzman, along with Marco Colonna, MD, the Robert Rock Belliveau, MD, Professor of Pathology and Immunology, and others have shown that when TREM2 is absent, the immune cells can't generate the energy they need to limit the spread of amyloid beta plaques.
Knowing that tau also plays a key role in the development of Alzheimer's, Holtzman, graduate student Cheryl Leyns and Jason Ulrich, PhD, an assistant professor of neurology, decided to investigate the effect of TREM2 on tau.
The researchers turned to genetically modified mice that carry a mutant form of human tau prone to forming toxic tangles. They snipped out the TREM2 gene in some of the mice so that all the mice developed tau tangles but only some of them also had the TREM2 protein in their microglia.
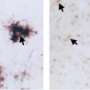
At 9 months of age, the brains of mice with tau tangles and TREM2 had visibly shrunk, particularly in areas important for memory. There was significantly less damage in the mice without TREM2.
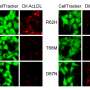
To their surprise, the researchers discovered there was no significant difference in the amount of tau tangles in the two groups of mice. Instead, the key difference seemed to lie in how their immune cells responded to the tau tangles. The microglia in mice with TREM2 were active, releasing compounds that in some circumstances help fight disease, but in this case primarily injured and killed nearby neurons. The microglia in mice without TREM2 were much less active, and their neurons were relatively spared.
"Once we started seeing the difference in the microglial activation, we started to understand that this damage doesn't have to do with tau aggregation necessarily, but with the immune system's response to the aggregation," said Leyns, the study's co-first author.
The findings suggest that the same immune cells can be involved in both protecting against and promoting neurological damage in Alzheimer's.
"It looks like microglial activation might have very different roles in different settings," Holtzman said. "Amyloid seems to set off the disease. You need these amyloid-related changes to even get the disease in the first place. Maybe that's why when you have less TREM2 function - and therefore microglial function - you're at higher risk of developing Alzheimer's. Less TREM2 function exacerbates amyloid-related injury. But then once the disease progresses and you start to have tau aggregation, it seems that activated microglia become harmful."
For years, doctors have tried to prevent or treat Alzheimer's disease by targeting amyloid plaques or tau tangles, but no therapy has yet been proven effective. When TREM2 was identified as a major risk factor four years ago, scientists seized on it as a fresh way of thinking about - and tackling - the disease.
These findings indicate that attempting to treat Alzheimer's by targeting TREM2 function and microglial activation may be a thornier problem than anyone had suspected.
"You might want to activate microglia early on, when people are just beginning to collect amyloid," Holtzman said. "If they're already developing symptoms, then they're later in the disease process, so you'd probably want to suppress microglia. However, these ideas would need to be thoroughly tested in animal models before we start talking about taking it into people."
More information: Leyns C, Ulrich J, Finn M, Stewart F, Koscal L, Remolina Serrano J, Robinson G, Anderson E, Colonna M, Holtzman DM. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proceedings of the National Academy of Sciences. Week of Oct. 9, 2017. www.pnas.org/cgi/doi/10.1073/pnas.1710311114