Molecular pathway offers treatment targets for pulmonary fibrosis, related conditions
. Credit: Wikipedia/CC BY-SA 3.0")
A study led by investigators at Massachusetts General Hospital (MGH) and University Health Network (UHN) in Toronto has identified a molecular pathway that appears to be critical to the development of fibrosis - scarring and excessive tissue deposition that result from abnormal healing responses and can compromise the function of vital organs. In their report that has received advance online publication in Nature Medicine, the researchers describe a series of molecular interactions that are essential for the induction of lung and skin fibrosis in a mouse model and appear to be active in human fibrotic lung disease.
"The ability of organs to regenerate following injury declines with age, so aberrant healing in response to chronic tissue injury in aged individuals can result in fibrosis - which contributes to 45 percent of deaths in the developing world," explains David Lagares, PhD, director of the Matrix and Mechanobiology Program at the MGH Fibrosis Research Center, co-corresponding author of the report. "Fibrosis is a component of chronic kidney disease, liver cirrhosis and pulmonary fibrosis; and the pathway we have identified could be a therapeutic target for halting the process and for restoring organ function."
Normal healing requires the interaction of many cell types - immune cells, stem cells, and the connective tissue cells called fibroblasts. Activated fibroblasts are called myofibroblasts and orchestrate many aspects of the healing process. But severe or repetitive injury can cause myofibroblasts to become hyperactive, setting off an out-of-control tissue buildup that leads to fibrosis. The molecular mediators underlying fibroblast activation are poorly understood, and their identification was a primary focus of the current study.
"We specifically wanted to identify factors that could control profibrotic activity without affecting normal wound healing," says co-corresponding author Mohit Kapoor, PhD, director of Arthritis Research in the Krembil Research Institute at UHN. "Identifying these factors will not only further enhance our understanding of the pathogenesis of fibrosis but will also provide new antifibrotic targets for multiple diseases."
The investigators first searched a gene expression database for genes that were upregulated in fibroblasts from patients with two important fibrotic diseases - idiopathic pulmonary fibrosis (IPF) and systemic sclerosis or scleroderma - compared with healthy individuals. One of the genes they identified codes for the cell membrane protein ephrin-B2, which binds to receptors on adjacent cells. During normal development ephrin-B2 regulates the formation and assembly of blood vessels, and its activity has been associated with the development of new blood vessels within and around malignant tumors.
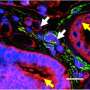
After confirming increased ephrin-B2 expression in a set of lung fibroblasts from patients with IPF, the team found that suppressing ephrin-B2 expression blocked the development of lung fibrosis in a mouse model. Further investigation revealed that the attraction of additional fibroblasts to sites of lung injury and their activation into myofibroblasts was set off when the portion of the ephrin-B2 protein that extends outside the cell membrane was clipped off by an enzyme called ADAM10, releasing a molecule called sEphrin-B2.
"Finding that this extracellular portion of ephrin-B2 was cleaved from the cell surface and secreted into the airspace following lung injury was totally unexpected and led us to hypothesize that this soluble form of ephrin-B2 may contribute to lung fibrosis," says Lagares, a principal investigator in the MGH Division of Pulmonary and Critical Care Medicine and faculty member at Harvard Medical School. "While ephrin-B2 typically interacts with receptors on different types of cells, both ephrin-B2 and its receptors are expressed on the surface of myofibroblasts, where their interaction usually suppresses the process leading to the overaccumulation of fibrotic tissue. In the novel mechanism we have discovered, the release of sEphrin-B2 prevents that inhibitory interaction and activates the receptors, leading to the amplification of tissue fibrosis."
In their mouse model of lung fibrosis, the inhibition of ADAM10 prevented the shedding of sEphrin-B2 and reduced fibrosis and death. Increased ADAM10-sEphrin-B2 signaling was also found in lung fibroblasts from IPF patients, and the enzyme's inhibition reduced sEphrin-B2 shedding in cells from both IPF patients and healthy volunteers. Suppression of ADAM10-sEphrin-B2 signaling also reduced the expression of several fibrosis-associated proteins in myofibroblasts from patients with IPF, and levels of sEphrin-B2 were higher in both lung fluid and plasma from patients than from healthy volunteers.
"Myofibroblasts are absent in healthy tissues, but their numbers and activity are markedly increased in fibrotic diseases," says Kapoor, a senior scientist at UHN. "Our identification of the ADAM10-sEphrin-B2 pathway as a promoter of myofibroblast activation gives us a number of attractive therapeutic targets for pulmonary and other fibrotic diseases." The team is continuing to investigate the fibrogenic program induced by ephrin-B2 signaling in activated myofibroblasts and testing the potential efficacy of neutralizing ephrin-B2 antibodies in tissue fibrosis.
A guiding force behind this work was the late Andrew Tager, MD, of the MGH Pulmonary and Critical Care Medicine, who founded what is now the Andrew Tager Fibrosis Research Center. "Andy was a giant in pulmonary fibrosis research and one of the most influential scientific minds in the field. His vision and insight were crucial to the discovery of the ADAM10-sEphrin-B2 pathway in pulmonary fibrosis" says Lagares. A co-author of this Nature Medicine paper and an associate professor of Medicine at Harvard Medical School, Tager passed away in August 2017.
More information: David Lagares et al, ADAM10-mediated ephrin-B2 shedding promotes myofibroblast activation and organ fibrosis, Nature Medicine (2017). DOI: 10.1038/nm.4419