New findings on autism-related disorder
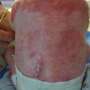
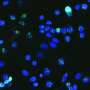
, cells lacking Adnp (right) displayed gross morphological changes and appeared to differentiate spontaneously as they started spreading out of characteristically densely packed ES cell colonies. Credit: Friedrich Miescher Institute for Biomedical Research")
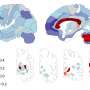
In a study published today in Nature, Marc Bühler and his group at the Friedrich Miescher Institute for Biomedical Research (FMI) have taken a major step forward in elucidating the mechanisms underlying a disorder known as Helsmoortel-Van der Aa syndrome. They clarify the role played by ADNP—a protein whose gene is mutated in patients with the syndrome—in the formation of tissues during embryonic development. The study provides important insights into a disorder that has been poorly understood to date, and for which no treatment is yet available.
Helsmoortel-Van der Aa syndrome is a rare condition, affecting an estimated one in 27,000 children. The mutations in the gene encoding ADNP (activity-dependent neuroprotective protein), which are responsible for this disorder were only identified in 2014. In addition, little is known about the disease mechanism – including the function of ADNP itself, except that it plays a role in the development of the brain. However, children with this syndrome may exhibit a wide variety of symptoms, including impairments of vision and the immune system, as well as motor and behavioral disturbances.
In a detailed study, the FMI group led by Marc Bühler has now clarified the function of ADNP, thus helping to explain how mutations in the ADNP gene can cause the various symptoms and suggesting a possible therapeutic approach.
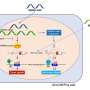
The group showed that, in mouse embryonic stem cells, ADNP interacts with DNA in a sequence-specific manner, and with two other proteins – HP1 and Chd4. Together, these proteins impair the accessibility of the DNA, which means that genes at these sites are no longer expressed. Bühler explains: "This previously unknown protein complex, which we call ChAHP, locally inactivates genes that would otherwise be active. If ChAHP is absent due to a mutation in ADNP, then all of a sudden genes will be transcribed, which should not yet be active." The scientists observed that, for example, instead of neuronal cells, other types of cells were formed as a result.
The inactivation depends solely on the DNA sequence and ADNP, and not – as in other cases – on further compaction of the DNA itself. Bühler comments: "This is a new inactivation mechanism, which is fascinating and will doubtless occupy us for some time."
Finally, the scientists noted that many of the mutations identified in patients with Helsmoortel-Van der Aa syndrome are nonsense mutations, leading to premature termination of protein synthesis – and therefore truncated proteins. "We showed in cells that reagents such as aminoglycosides which suppress this premature stop can restore the molecular function of ADNP. Patients could thus benefit from therapeutic agents which make it possible for these nonsense mutations to be ignored." This is of particular interest given that over 2000 diseases are caused by mutations that lead to premature termination of protein synthesis.
More information: Veronika Ostapcuk et al. Activity-dependent neuroprotective protein recruits HP1 and CHD4 to control lineage-specifying genes, Nature (2018). DOI: 10.1038/s41586-018-0153-8