Researchers look to worms for a new model of a peripheral nervous system disease

Studying transthyretin amyloidoses-a group of progressive nerve and cardiac degenerative diseases caused by the buildup of misfolded transthyretin (TTR) proteins in the body-has long been hampered by the lack of animal models of the disease. Mice, for instance, don't show the same symptoms as humans, even when misfolded TTR accumulates in their organs.
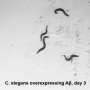
Now, scientists at Scripps Research have discovered that Caenorhabditis elegans, a nematode, or microscopic roundworm, develops similar nerve damage to human patients when their muscle cells are genetically engineered to produce TTR.
"This is really the first model that recapitulates what we see in humans both with regards to the molecular and cellular signatures of the disease, and the symptoms," says Sandra Encalada, Ph.D., Arlene and Arnold Goldstein Assistant Professor of Molecular Medicine at Scripps Research.
The new C. elegans model, which Encalada and her team described recently in the journal Proceedings of the National Academy of Sciences, has already let Scripps Research scientists make inroads into understanding how TTR proteins become misfolded and aggregate to cause disease in neurons.
In humans, TTR is produced and secreted by the liver, where sets of four copies of the protein assemble together into tetrameric TTR that's sent out into the bloodstream. In the blood, TTR normally binds and transports the thyroid hormone thyroxine, as well as vitamin A bound to retinol binding protein to deliver them throughout the body.
But there's a ticking clock: the four TTR copies also fall apart over time, and then, in some cases, change their conformation or shape and regroup or misassemble into larger aggregates that deposit in tissues. There is genetic and pharmacologic evidence that this process causes neurodegeneration.
People can suffer from a variety of diseases based on the sequence of TTR that misfolds and misassembles and depending on where misfolded TTR aggregates accumulate. In the two most common forms of TTR amyloid disease, the protein accumulates in the heart-causing cardiac symptoms-or in the nerves of the legs and arms-causing a peripheral neuropathy. While some people who develop these diseases have mutations in their TTR protein, making them more prone to aggregate, others have normal TTR that can also misfold and misassemble.
"We know quite a bit about the molecular dynamics of how TTR comes apart and how it creates aggregates," says Encalada. "But until now we didn't have any mechanism at the cellular level. How do heart or nerve cells degenerate when TTR aggregates?" Scientists working with dozens of rodent and fruit fly models have failed to replicate what is seen in humans with these conditions.
In an attempt to answer these questions, Encalada and her collaborators engineered C. elegans to produce TTR in their muscle cells. They then tested the bodies of the nematodes for the presence of TTR. The protein, they showed, was secreted out of the muscle cells and into the worms' body cavity. And just as in humans, the TTR broke down from tetramers and converted into misfolded and aggregated TTR molecules over the course of about a week.
When the researchers gave the nematodes a mutated version of TTR known to cause progressive peripheral neuropathy in humans, the worms showed abnormal growth of sensory nerve cells, and lost the ability to feel pain and temperature -the same impairments that are seen in humans. Moreover, when the worms were treated with drugs that ameliorate TTR peripheral neuropathy in humans, the worms showed dramatic improvement of the aforementioned degenerative phenotypes.
Encalada's team tracked where the TTR was going in the worms' bodies, and they found that tetramers of the protein secreted from the muscle, accumulated in the cells responsible for breaking down the body's waste. These cells, the researchers showed, were degrading TTR and preventing the production of toxic aggregates. Deleting these cells enhanced the aggregation of TTR and increased the percentage of animals that had signs of nerve disease, including loss of pain sensation, as observed in humans.
"The big picture is that we were able to modulate levels of TTR degradation without touching neurons or the muscle cells producing TTR," says Encalada. "In humans, being able to tweak levels of TTR degradation could act as a means of stopping TTR toxicity."
More work is needed to determine whether the observations in C. elegans can be recapitulated in humans regarding the ability of specific cells to degrade TTR, but Encalada is hopeful that the new animal models will fuel more research into TTR-linked diseases. In addition, she says, the overall findings on the link between protein degradation and nerve toxicity could translate to other neurodegenerative diseases, including Alzheimer's.
More information: Kayalvizhi Madhivanan et al, Cellular clearance of circulating transthyretin decreases cell-nonautonomous proteotoxicity in Caenorhabditis elegans, Proceedings of the National Academy of Sciences (2018). DOI: 10.1073/pnas.1801117115