'Crosstalk' between genes promotes brain inflammation in Alzheimer's

A new study by scientists at Massachusetts General Hospital (MGH) offers clues about how to prevent inflammation of brain tissue, which promotes Alzheimer's disease (AD). The findings of this study online now and appearing in the September 4, 2019 print issue of the journal Neuron, could contribute to the development of new therapies for AD.
It's known that the brains of people with AD fill with deposits of damaged nerve cells and other proteins, known as amyloid plaques, as well as tangled formations of proteins called tau. "But if you just have plaques and tangles alone, you probably won't develop Alzheimer's disease for a long time, if at all," says neuroscientist Rudolph E. Tanzi, Ph.D., director of the Genetics and Aging Research Unit at MGH, and senior author of the Neuron study. Rather, explains Tanzi, it's the inflammation that occurs in response to plaques and tangles, or neuroinflammation, that is the primary killer of neurons, which leads to cognitive decline.
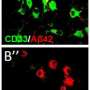
Tanzi's lab discovered the first gene associated with neuroinflammation in AD, known as CD33, in 2008. CD33 carries the genetic code for receptors found on microglia cells, which normally act as one of the brain's housekeepers, clearing away neurological debris, including plaques and tangles. In 2013, Tanzi and colleagues published their discovery that CD33 influences the activity of microglia: When the gene is highly expressed, microglia turn from housekeepers to neuron killers, sparking neuroinflammation.
Meanwhile, other investigators identified another gene, TREM2, which has the opposite effect of CD33: It shuts down microglia's capacity to promote neuroinflammation. In other words, says Tanzi, CD33 is the "on" switch for neuroinflammation, while TREM2 acts like an "off" switch. "The Holy Grail in this field has been to discover how to turn off neuroinflammation in microglia," says Tanzi.
In their most recent inquiry, Tanzi, neuroscientist Ana Griciuc, Ph.D., and their colleagues set out to discover how CD33 and TREM2 interact, and what role that "crosstalk" might play in neuroinflammation and the origin of AD. To do that, they posed a question: What happens when these critically important genes are silenced—individually and simultaneously?
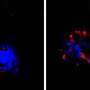
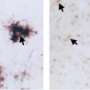
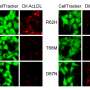
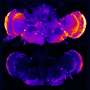
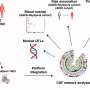
To find answers, Tanzi and his team studied laboratory mice specially bred to have brain changes and behavior consistent with AD. The team began by observing and testing a strain of AD mice that had their CD33 genes turned off. They discovered that these mice had reduced levels of amyloid plaque in their brains and performed better than other AD mice on tests of learning and memory, such as finding their way in a maze. However, when mice had both CD33 and TREM2 silenced, the brain and behavior benefits disappeared—which also happened when only a single TREM2 gene was quieted. "That tells us that TREM2 is working downstream of CD33 to control neuroinflammation," says Tanzi. That theory was bolstered by sequencing of microglia RNA, which indicated that both CD33 and TREM2 regulate neuroinflammation by increasing or decreasing activity of an immune cell called IL-1 beta and the cell receptor IL-1RN.
"We are increasingly realizing that to help Alzheimer's patients, it is most critical to stop the massive brain nerve cell death that is caused by neuroinflammation," says Tanzi. "We now see that the CD33 and TREM2 genes are the best drug targets for achieving this goal."
More information: Ana Griciuc et al, TREM2 Acts Downstream of CD33 in Modulating Microglial Pathology in Alzheimer's Disease, Neuron (2019). DOI: 10.1016/j.neuron.2019.06.010