New clues about the origins of familial forms of amyotrophic lateral sclerosis
. Credit: Aline Brasil, Elis Eleutherio, and Tiago Outeiro")
A team led by Brazilian researcher Elis Eleutherio, professor at the Federal University of Rio de Janeiro, in partnership with Tiago Outeiro, at University of Goettingen, Germany, made important progress in understanding the conformation and accumulation of certain proteins involved in amyotrophic lateral sclerosis (ALS).
"We believe protein accumulation is an important hallmark of ALS, and we still do not understand why the protein misbehaves and aggregates during the disease," explains Prof. Eleutherio.
ALS is a progressive and devastating neurodegenerative disorder affecting 1 to 3 individuals in 100,000, and it is more prevalent in people between 55-75 years of age. The disease affects primarily a population of neurons known as 'motor neurons.' Patients suffer from irreversible motor paralysis, and become incapable of speaking, swallowing, or breathing as the disease progresses.
Most ALS cases are sporadic, with no defined genetic origin, and only the minority is familial, with known associated genetic alterations. Certain familial forms of ALS (fALS) are associated with genetic alterations in the gene encoding for a protein known as superoxide dismutase 1 (Sod1), that cause alterations in the folding and function of the protein.
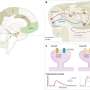
The study, published in the journal Proceedings of the National Academy of Sciences (PNAS), allowed scientists to understand the interaction between the normal and the mutant protein, which causes alteration of protein accumulation in the cell, but also impairs the function of the Sod1 protein, thus contributing to the development of the disease. For the group, this discovery opens new perspectives for the treatment of ALS.
Sod1 is a protein that plays a role, among others, in the protection against oxidative damage in our cells. In some ALS cases, altered Sod1 protein accumulates inside neuronal cells and, researchers believe, causes damage to the neurons, leading to their death. Importantly, normal Sod1 proteins present in sporadic cases of ALS, can also misfold and accumulate, suggesting this is a central problem in ALS.
In the study, the researchers used simple experimental models, such as bakery yeast used to make beer, wine and bread, and human cells, in order to better understand the context of protein aggregation in the disease. They also used a strategy that mimics the genetic context of fALS, where most patients carry one copy of the normal Sod1 protein, and one copy carrying a genetic alteration. "In patients, we think that the presence of a mutant copy of Sod1 alters the behavior of the normal copy," explains Dr. Aline Brasil, the first author of the study.
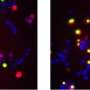
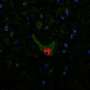
"By taking advantage of novel genetic manipulation tools, and powerful molecular imaging approaches that enable the direct visualization of protein complexes in the cell (a technique known as BiFC), we were able to detect 'hetero-complexes' formed by normal and abnormal (mutant) Sod1 protein," said Prof. Outeiro, leader of the German team that participated in the study.
The research opens novel perspectives for therapeutic intervention that the authors hope to continue to explore in the near future, such as the specific removal of mutant Sod1 protein.
"In a time when the scientific and educational system in Brazil suffers from uncertainty, it is important to demonstrate that we can be competitive and conduct research that will contribute to society and may, ultimately, help change the lives of those affected by such devastating diseases," Prof. Eleutherio concludes.
More information: Aline de Araújo Brasil el al., "Characterization of the activity, aggregation, and toxicity of heterodimers of WT and ALS-associated mutant Sod1," PNAS (2019). www.pnas.org/cgi/doi/10.1073/pnas.1902483116