Pancreatic β cell-derived exosomal miR-29 family enhances hepatic glucose output

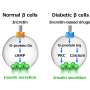
In a new study published in Journal of Extracellular Vesicles, Chen-Yu Zhang's group at Nanjing University, School of Life Sciences, and Antonio Vidal-Puig's group at University of Cambridge report that pancreatic β cells secrete miR-29 family members (miR-29s) via exosomes in response to high levels of free fatty acids (FFAs). Theses β cell-derived exosomal miR-29s regulate glucose homeostasis through their manipulations on glucose output in liver.
Previously, Chen-Yu Zhang's group identified extracellular miRNA as a new form of cell-to-cell communication. They are among the first that reported the selective secretion of miRNAs under different physiological or pathological states; also, the uptake and function of secreted miRNAs in recipient cells. In the past decade, intensive studies have revealed the role of extracellular miRNAs in a range of biological processes. Thus, as a newly-emerged secretory, more insightful studies are need to further reveal its relevance to more physiological progresses and diseases. Pancreatic islet has long been identified as critical secretory tissue in term of its role on maintaining glucose homeostasis by releasing conventional hormones, such as glucagon and insulin. Since pancreatic islet is a classic secretory organ, study to identify its secreted miRNAs and their functional implications in the regulation of glucose homeostasis is very needed.
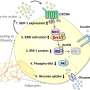
In the current study, they show that high levels of FFAs can selectively induce the secretion of miR-29s from cultured pancreatic β cells. In vivo study revealed both physiological (fasting)-and pathological (obese)-associated high levels of FFAs induce the secretion of miR-29s from pancreatic β cells. Likewise, miR-29s are also increased in the plasma of obese human compared to normal human. Intriguingly, they discovered that exosomal miR-29s administrated intravenously can cause impaired insulin sensitivity. Next, they used a combination of genetic modification animals to further confirm the functional role of secreted miR-29s. First, mice overexpressed miR-29s in β cells show insulin resistance and an enhanced hepatic glucose output, indicating β cell-derived miR-29s may target liver and impact hepatic insulin action. To confirm this, a distinguishable mutant miR-29a (with four nucleotides mutated in non-seed region) was overexpressed in pancreatic β cells of mice. Consistently, these mice show impaired insulin sensitivity. By tracking the distribution of mutant miR-29a, they found β cell-derived mutant miR-29a can be taken up by liver. Mechanism dissections revealed that β cell-derived miR-29s negatively regulate insulin signaling pathway via targeting p85 α (regulatory subunit of PI3K), and subsequently attenuate the suppression of insulin on glucose output. Finally, miR-29s deficiency in β cell significantly improves the insulin sensitivity in mice fed on HFD. This study not only identifies a new secretory factor from pancreatic β cells, but also elucidates an alternative mechanism that underlies β cell-controlled glucose homeostasis.
This work is important for the following reasons:
- We demonstrate pancreatic islets secrete not only canonical hormones, but also exosomal miRNAs, which is largely extended our understanding of islet function;
- We identify a new secretory that play a role in regulation of glucose homeostasis.
- We reveal a mechanism underlying obesity/FFAs-induced insulin resistance and type 2 diabetes. Obesity or high levels of FFA may not only directly affect liver, skeletal muscle and white adipose tissue and cause insulin resistance, but regulate exogenous secreted miRNA to indirectly result in pathophysiology as well. More importantly, the secreted miR-29s is increased prior to the onset of insulin resistance in ob/ob mice, indicating secreted miR-29s may be the factors that initiate the development of insulin resistance.
- We provide further evidence for the selectivity of secreted miRNAs under certain physiological or pathological context.
More information: Jing Li et al, Pancreatic β cells control glucose homeostasis via the secretion of exosomal miR‐29 family, Journal of Extracellular Vesicles (2021). DOI: 10.1002/jev2.12055