Drug discovery approach reveals why drugs for cystic fibrosis fall short

To cure rare genetic diseases, from cystic fibrosis to Niemann-Pick, scientists at Scripps Research have turned to a computational approach usually used to pinpoint the best spot for an oil well. By using the method to analyze the spatial relationships between different variants of a protein—instead of the relationships between test wells across a landscape—the researchers can obtain valuable information on how disease affects a protein's underlying shape and how drugs can restore that shape to normal.
The new method, detailed today in the journal Structure, requires only a handful of gene sequences, collected from people with disease. Then, it determines how the structure of each corresponding variant protein is associated with its function, and how this functional structure can affect pathology and be repaired by therapeutics. To show its utility, the Scripps Research team used the method to show why existing drugs for cystic fibrosis fall short of curing the disease.
"This is an important step forward for treating rare diseases," says senior author William Balch, Ph.D., professor of Molecular Medicine at Scripps Research. "The fact that we can get so much information from a few gene sequences is really unprecedented."
Studies on inherited diseases often rely on techniques that determine the precise three-dimensional shape of a protein affected by disease. But genetic diseases can be caused by dozens—or even hundreds or thousands—of different changes to the same gene, called variants. Some of these variants destabilize or change the protein shape in ways that make isolating the protein for further investigation much more difficult than usual.
Balch, with Scripps Research senior staff scientist Chao Wang and staff scientist Frédéric Anglés, instead wanted to use natural variation to their advantage. For most genes in the human genome, numerous variants exist in the human population; some of these variants cause disease and others have little impact on biology and go unnoticed. So the group developed a method called variation-capture (VarC) mapping to analyze this natural array of gene sequences and determine the mechanism by which they each changed a protein's structure to cause disease.
Balch's group integrated a handful of machine learning and statistical tools into VarC, including the methods that oil companies use to draw inferences about the location of an oil reservoir using only a small number of test wells. With only a few gene sequences this let the researchers determine the most likely structural mechanisms driving function for each variant leading to disease, as well as model how drugs impacted those structural functions.
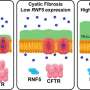
In the example of cystic fibrosis, the disease is caused by genetic variants in the cystic fibrosis transmembrane conductance regulator (CFTR), leading to a buildup of mucus in the lungs. More than 2,000 variants of the CFTR gene have been identified and described in a patient database. Researchers knew that many of these variants had very different effects on the CFTR protein, but it has been difficult to compare and contrast these variants to guide how patients with different variants should be treated differently in the clinic.
"When you want to treat patients, you really have to appreciate that different therapeutics might target different variants in completely different ways, and that's why our approach that looks at many different variants all at once is so powerful," says Wang. "Our approach not only reveals how these variants contribute to each patient's biology, but also connects them in a way that each variant can inform how to manage the others."
The researchers input the information of about 60 genetic variants found in the cystic fibrosis population into their VarC program. The computational analysis captured how each amino acid residue talks to every other residue to generate function, and revealed that most of the cystic fibrosis patients had the same net effect on the protein: an unstable inner core.
When the Scripps Research team then used the program to model how existing cystic fibrosis drugs impacted the structures, they discovered that—although the drugs impact the CFTR protein in different ways—none of them effectively stabilized the protein's inner core hidden from view, like the location of an oil reservoir in a complex landscape that is revealed by test wells.
Now that Balch and his colleagues better understands the structural deficiencies in CFTR in cystic fibrosis patients, they say that the job of developing an effective drug to fix it is much easier. Potential compounds can be modeled in advance of lab experiments for their effect on the inner core of the CFTR protein.
"In most drug discovery, you throw thousands of compounds at a protein and see which ones change it, often without fully understanding the mechanism," says Balch. "To fix a thing, you must first understand the problem."
And Balch adds that cystic fibrosis isn't the only disease likely to be solved with their VarC approach. Any genetic disease can be analyzed in the same way, using knowledge of patient variants found in the population along with the information on symptoms triggered by each variant.
"We really think we can do this for any protein out there," says Balch. "It's a fast track toward drug discovery for rare diseases that have been very hard and slow to study in the past."
Already, his team is applying the method to other rare genetic diseases, as well as pursuing new drugs to treat cystic fibrosis.
More information: Chao Wang et al, Triangulating variation in the population to define mechanisms for precision management of genetic disease, Structure (2022). DOI: 10.1016/j.str.2022.05.011