This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
trusted source
proofread
Conquering chronic lung infection in cystic fibrosis patients
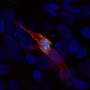
 surrounded by alginate, an amorphous substance. Credit: Peter Gilligan, Ph.D.")
Cystic fibrosis (CF) is one of the most common genetic disorders in the world. The disease infects nearly 70,000 people worldwide and approximately one in 30 Americans is a carrier. CF is caused by mutations in the human cystic fibrosis transmembrane regulator (CFTR) gene, and more than 2,000 mutations have been characterized in people with CF (PwCF). CFTR mutations cause physiologic changes that result in chronic lung infections, the major culprit of which is an unusual phenotype of Pseudomonas aeruginosa called "mucoid."
For decades, chronic lung infection has been responsible either directly or indirectly for 70% of pre-mature deaths in PwCF. However, the seminal discovery of the CFTR gene in 1989 led to the development of small molecules that can modulate the activity of CFTR. The 2019 approval of the small molecule-based medication Trikafta has provided hope to PwCF and their families for a much-prolonged life span. This oral drug modifies the activity of the aberrant CFTR proteins in 90% of PwCF. It is given twice daily either as granules for children 2–6 years of age, or as tablets for those older than 6. It is well tolerated but can have liver toxicity and a wide array of drug-drug interactions.
Two scientists, Peter Gilligan, Ph.D., former Director of the Clinical Microbiology-Immunology Laboratories at the University of North Carolina (UNC) Hospitals and Rhianna Lee, Ph.D., who works in the Marsico Lung Institute/UNC Cystic Fibrosis Center, describe the battle to fight CF lung disease and the remarkable advances science has made in this fight.
Why is CF of particular and personal interest to you?
Gilligan: As I approach my 50th college reunion, it is a time of reflection. Any reflection of that time must include my roommate of four years, Leon. Unknown to me, Leon was living on borrowed time when I met him, for he had cystic fibrosis. In 1969, the median life expectancy of a PwCF was 14 years. Leon's dream was to graduate from Holy Cross, which we did proudly in 1973. He passed away four years later. His life and experience battling CF have provided inspiration for my scientific journey, which continues to this day.
Lee: I was 8 years old when I first heard the words cystic fibrosis. The diagnosis was given as the reason for my brother's sudden hospitalization. He was 3 months old at the time, but those words have defined each moment of his life—and mine—ever since. At the time, I knew very little about the disease or its implications, only that my parents were fearful of what CF could mean for my brother. I sat with him through countless breathing treatments, encouraged him during hospital stays and, all the while, gleaned hope from the impact that research was having on his prognosis. By the age of 12, I decided to one day be a part of the research that was actively changing my brother's life.
Pathophysiology of CF lung disease
The airway epithelium serves as a vital barrier between the body and the outside world, protecting against uptake of inhaled particles, chemicals, allergens and pathogens. Inhaled materials are trapped in secreted mucus and removed from the airways through a process known as mucociliary clearance (MCC), in which motile cilia beat in a coordinated fashion to move particulates up and out of the respiratory tract. Proper hydration of the airway epithelium and the overlying mucus layer is key to productive MCC.
The CFTR gene encodes for an epithelial ion channel, which conducts chloride (Cl-) and bicarbonate (HCO3) out of epithelial cells and into the airway lumen. Water follows these molecules, thus hydrating the airway surface layer (ASL) and mucus layer covering the airway epithelium. In CF, absent or dysfunctional CFTR leads to a dehydrated ASL and buildup of a thick, sticky mucus, which cannot be transported by MCC. Pathogens become trapped in these abnormal lung secretions, leading to chronic infection and persistent inflammation.
The major pathogen responsible for chronic lung infection in CF patients is mucoid Pseudomonas aeruginosa (PA), which grows as microcolonies in the CF airway. These microcolonies are composed of gram-negative bacilli encased in the polysaccharide, alginate, and aggregated in biofilms. Alginate gives colonies their mucoid appearance.
Mucoid PA is refractory to MCC, phagocytosis and antimicrobial therapy allowing the establishment of chronic infection, often leading to chronic inflammation. The combination causes progressive damage to the lung architecture, resulting in ever-worsening lung function. Over a period of years, this eventually leads to either lung transplantation or death. Other important pathogens encountered in chronic CF lung infections include Staphylococcus aureus (especially methicillin resistant strains), Burkholderia cepacia complex, Mycobacterium abscessus complex and the fungus Aspergillus fumigatus. PwCF also experience acute infections with respiratory viruses such as Respiratory syncytial virus RSV and influenza, although the impact of viruses on chronic bacterial infections is less certain. Interestingly, COVID-19 has not been a significant cause of morbidity and mortality in PwCF, with mild disease being the norm.
Treatment of CF lung disease
Small molecule CFTR modulators
The landscape of CF treatment shifted dramatically with the development of small molecule CFTR modulators, which address the underlying cause of disease by correcting protein folding, trafficking, function or stability. This results in improved MCC, limiting chronic infection, especially due to mucoid PA. The first CFTR modulator, Kalydeco, was approved in 2012 for PwCF carrying at least one copy of the G551D variant. Kalydeco is highly effective, rescuing about 11% of pulmonary function—measured as the forced expired volume in one second, or FEV1. However, only ~4% of the CF population was eligible for this treatment, based on the genotype criteria that were initially established.
The next big breakthrough occurred in 2015, when Orkambi was approved for PwCF carrying two copies of the F508del variant. F508del is the most common CFTR variant, so this covered about 50% of the CF population. However, Orkambi was less effective than Kalydeco, rescuing only ~3% of pulmonary function in clinical trials in 2017. Through in vitro studies of CFTR function, Kalydeco eligibility was expanded to include 23 relatively rare CFTR variants, extending modulator treatment to another 6% of the population.
Then in 2018, Symdeco was approved, showing marginal improvements over Orkambi. Finally in 2019, the blockbuster drug, Trikafta, was approved for PwCF carrying at least one copy of the F508del, the most frequent variant covering ~80% of the CF population. Moreover, Trikafta is the most effective CFTR modulator developed to date, correcting ~14% of pulmonary function in trials.
Overall, CFTR modulators have been extremely successful in extending life expectancy and improving the quality of life for 90% of PwCF, but developing therapies for the remaining 10% has proven challenging. This group harbors a wide range of exceptionally rare CFTR variants. In fact, more than 1,200 CFTR variants are carried by five or fewer people worldwide. For these individuals, a well-powered clinical trial will never be possible. This group also includes CFTR variants that cause premature termination codons, which produce little to no CFTR protein and, thus, little to no drug target. As the field shifts its focus toward the long list of rare, untreated CFTR variants, it becomes clear that extending life-changing treatment to all PwCF will require a new approach to drug design and development.
Prior to CFTR modulators, the care of CF patients targeted the mobilization of respiratory airway secretions, the eradication of bacterial pathogens and treatment of pulmonary exacerbations.
Mobilization of airway secretions
Because the thick, mucous secretions found in CF airways are an ideal habitat for certain bacteria, such as P. aeruginosa and S. aureus, mobilization and clearance of airway secretions has been an important treatment for PwCF. For many years, a standard treatment was to have a family member percuss the back and chest of the PwCF to mobilize the thick, sticky mucous secretions. A more modern approach to this treatment requires the patient to wear an oscillating vest that mobilizes the secretions. After chest physiotherapy, the patient typically uses a nebulizer to deliver treatments including DNAse and hypertonic saline making mucous less sticky, more watery and easier to expectorate.
Antimicrobial eradication of pseudomonas aeruginosa
To prevent the establishment of chronic PA infection, eradication therapy using aerosolized antimicrobials has become a standard of care in CF patients, with tobramycin being the most used antimicrobial. However, eradication is somewhat of a misnomer, since the primary goal of this strategy is to delay the onset of chronic infection. The decline in the prevalence of P. aeruginosa in PwCF suggests that this approach, along with genetic modifiers, has delayed the establishment of infection.
Treatment of pulmonary exacerbation
The length and quality of life in PwCF is based on the frequency of pulmonary exacerbation. It is an acute manifestation of chronic lung infections characterized by fever; increased cough, sputum production and respiratory rate; weight loss and declining respiratory function, as measured by spirometry. People with frequent exacerbations tend to have more frequent hospitalizations, a poorer quality of life and shorter lifespans. Exacerbations are typically treated with two or three intravenous (IV) antimicrobials for 14 days. This treatment does not "cure" the infection. Rather it reduces clinical symptoms and may restore some lung function. However once chronic infection is established, damage to lung architecture is relentless, eventually leading to lung transplantation or death, over a period of years to decades.
When will there be a cure for CF lung disease?
The detection of the CFTR gene in 1989 raised hope that gene therapy could be successful in reversing the genetic defect in this gene. A 1995 proof of concept clinical trial of the transfer of a normal CFTR gene into nasal epithelial cells using an adenovirus vector was unsuccessful. Multiple trials have followed with none showing significant success.
Double lung transplantation is a proven "cure" for CF lung disease. First done extensively in the 1990s, the median life expectancy of CF transplant recipients has steadily increased, reaching 9.5 years by 2018. However, the 2021 U.S. CF patient registry reports that complication of lung transplantation was second only to chronic lung infection as a cause of death. With the wide availability of triple CFTR modulators, the number of CF lung transplants has plummeted. Clearly CFTR modulators are judged to be better options than CF lung transplantation in the care of most PwCF.
The success of highly effective CFTR modulators highlights the importance of correcting the underlying cause of CF. While modulators act at the protein level, a long-term cure will require looking upstream to correct the genetic defect at the DNA level. Theoretically, this could be achieved by one of three approaches:
- In vivo editing of the mutant CFTR sequence (i.e., gene editing).
- Delivery and integration of a normal CFTR sequence, (i.e., gene therapy).
- Delivery and integration of cells carrying a normal CFTR sequence (i.e., cell therapy).
The largest barrier facing each of the three approaches is delivery.
The airway epithelium serves a vital protective role by keeping foreign particles out of the human body. Unfortunately, these mechanisms cannot distinguish between harmful inhaled particles and those that are therapeutically delivered. Thus, airway mucus and MCC represent a major barrier to effective delivery of gene editing, gene therapy or cell therapy vectors. Methods to overcome the airway epithelial barrier safely and temporarily must be developed for successful gene correction or replacement.
Lee: Since my brother's diagnosis in 2001 to now, life expectancy for CF has increased by nearly 20 years. This advancement can be credited to the development of better airway clearance techniques, antimicrobial treatments and highly effective CFTR modulators. The latter demands particular recognition. Trikafta undoubtedly changed the trajectory of my brother's life. Though stories like his are cause for great celebration, the sobering reality is that many PwCF are still waiting for their breakthrough therapies. Their continued fight against CF and the hope provided by recent breakthroughs inspires me and many others to continue working toward a cure for CF.