This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
Fusion oncoprotein forces cell fates toward rhabdomyosarcoma
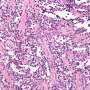
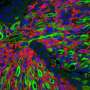
 from aP2-Cre;R26-tdTom sternocleidomastoid (SCM) and gastrocnemius (gastroc) following cardiotoxin (CTX) injury. Laminin (green), tdTomato(red), DAPI nuclear stain (blue) (n = 3). Scale bar = 100 μm. b Representative IF of SCM and quadricep femoralis hindlimb (HL). PAX7 (white), laminin (green), tdTomato (red), DAPI (blue). This experiment was repeated 3 times with similar results. Scale bar = 10 µm. c Representative IF of myogenic differentiation from ACP SCM and HL. Myosin heavy chain (MyHC, green), tdTomato (red), DAPI (blue)(n = 3, 100+ nuclei counted/replicate) Scale bar = 50 μm. Flow cytometry analysis of muscle stem cells by (MAC1/CD45/TER119) and SCA1 negative, β1-Integrin and CXCR4-positive, and Tom+ cells from d Pax7-CreERT2;R26-tdTom muscle - (n = 3 biological replicates) (P = 0.0002) and e Flow cytometry from ACP SCM and HL (n = 3 biological replicates) (P = 0.0314). f Representative IF from CTX injured Pax7-CreERT2;R26-tdTom gastroc (n = 2), and ACP SCM (n = 2) and gastrc (n = 2). Laminin (green), tdTomato (red), DAPI (blue). Scale bar = 100 μm. P values determined by Student’s t-test (unpaired, two-tailed); *P < 0.05 and ***P < 0.001. Data represented as mean+/- SEM. Source data are provided as a Source Data file. Credit: Nature Communications (2023). DOI: 10.1038/s41467-023-43044-1")
Pediatric rhabdomyosarcoma is a type of cancerous tumor that arises in the soft tissue, such as muscles, though the origin of one of the most aggressive subtypes is unclear.
Scientists at St. Jude Children's Research Hospital showed in proof-of-concept models that expressing one mutated hybrid "fusion" oncoprotein in either muscle or blood vessel (endothelial) cells produced these aggressive tumors, which were molecularly identical. The findings, which have implications for treating aggressive rhabdomyosarcoma, were published today in Nature Communications.
Rhabdomyosarcoma expressing a hybrid protein from the fusion of two genes, PAX3 and FOXO1, is referred to as fusion-positive. Pediatric fusion-positive rhabdomyosarcoma has a dismal prognosis.
Rhabdomyosarcoma cells appear muscle-like; however, fusion-positive tumors can form in areas without skeletal muscle. Not knowing where the cancerous fusion-positive cells come from can limit research and treatment options. This St. Jude study generated models to find where and how these fusion-positive tumors arise, ultimately providing clues for ways to stop this cancer.
"There's a subset of these patients with rhabdomyosarcoma that we don't know where the tumors come from," said corresponding author Mark Hatley, M.D., Ph.D., St. Jude Department of Oncology. "Our new models will allow us to shed light on their origin so that we can better treat them."
Fusion overrides fate to cause rhabdomyosarcoma
Fusion-positive rhabdomyosarcoma appears diffuse within the body, looking similar to metastatic disease. Historically, researchers attempted to find a primary muscle tumor to find a more effective way to counter this subtype by identifying its cell of origin. All such efforts to date have been unsuccessful.
Hatley's lab took a different approach. Previously, they showed fusion-negative rhabdomyosarcoma could come from non-muscle cells. The researchers wondered if that was also possible for the more aggressive subtype.
Therefore, they created a mouse model expressing the PAX3-FOXO1 fusion oncoprotein in blood vessel cells. They compared this to a previously established mouse model of fusion-positive rhabdomyosarcoma that arises from muscle cells. These mouse models enabled the researchers to see if they could find traces of the original cell type when presented with fusion-positive tumors from each model, thus distinguishing their origins and identifying potential vulnerabilities.
"We showed this fusion oncoprotein is such a potent rhabdomyosarcoma driver, it can overwrite a cell's identity," said co-author Brian Abraham, Ph.D., St. Jude Department of Computational Biology, whose lab performed much of the bioinformatics analysis in collaboration with the St. Jude Center for Applied Bioinformatics. "It overrode any sort of normal differentiation processes, in either muscle or endothelial cells, and forced those cells to become tumor-like."
"When we compared the tumors that develop from muscle or endothelial cells, they were indistinguishable," Hatley said. "We performed detailed analyses, including gene expression, chromatin architecture, and enhancer accessibility. Despite how hard we looked, we could not find a single distinguishing feature that told us that the tumor from the endothelial cells came from endothelial progenitors and not muscle cells."
Finding faults in fusion-positive rhabdomyosarcoma
The study indicates that it may not be possible to find the cellular origin of these cancerous cells and identify potential drug targets using those methods. Instead, focusing on the fusion oncoprotein's effects may be a better way to identify potential therapeutic interventions.
"The problem is that the fusion oncoprotein is a transcription factor, a protein which regulates gene expression but is currently undruggable," Abraham said. "We needed a way to look for genes targeted by the oncoprotein that are driving cancer cell identity and develop strategies to affect them instead."
The Hatley lab, therefore, created a model to look at the genetic and epigenetic effects of the fusion oncoprotein more closely. They exploited the oncoprotein's strength as a cancer driver in induced pluripotent stem cells. These cells have the capability of differentiating into any cell type. The scientists first differentiated the cells into endothelial cells and then added the fusion oncoprotein. This in vitro system allows them to look even closer at what happens when these cells become tumor-like.
Abraham's lab found that the fusion oncoprotein was still acting as a powerful activator of muscle cell fate in the system. The aberrant protein bound to DNA regions that enhanced expression of genes well-known to be major regulators of muscle cell development, such as MYOD1. These findings serve as a proof of concept that the system can find downstream targets of the oncoprotein and increase confidence in any future novel targets found by the method.
"This in vitro system reprograms blood vessel cell progenitors into a pediatric tumor that gives us a novel window to dissect the fundamental basis of rhabdomyosarcomas' cell fate," Hatley said. "In patients, we know these tumors are unique because those children do worse. This system has the potential to increase our understanding of those tumors, especially the fusion oncoprotein's downstream targets, and give us new avenues for treatment."
More information: Madeline B. Searcy et al, PAX3-FOXO1 dictates myogenic reprogramming and rhabdomyosarcoma identity in endothelial progenitors, Nature Communications (2023). DOI: 10.1038/s41467-023-43044-1