New cancer driver found: Monoclonal antibody therapy stops tumor growth in mice
 that reach out to neighboring cells. These connections initiate a process that creates cell-to-cell bindings that mimic attachment to the extracellular matrix and allow these cells to survive when they shouldn't, turning them into cancer cells. Credit: Elledge lab")
(Medical Xpress)—Approximately 90 percent of cancers start within tissues that form the inner linings of various organs. Decades of accumulated genetic mutations can, on occasion, induce cells specialized for growth in one-cell deep sheets to form disordered clumps that eventually become tumors.
New work from the lab of Stephen Elledge, Gregor Mendel Professor of Genetics and professor of medicine at Harvard Medical School, has found a genetic switch that changes these cells into cells with distinct cancerous properties. The group has also identified a way to flip this switch off to halt tumor growth.
"We've demonstrated a potential new therapy in mice that disrupts the activity of this new oncogene and impacts tumor growth in a significant way," said Elledge, who is also a professor of medicine at Brigham and Women's Hospital. "This therapy is in line with other biological therapies out there, so it could be used on people."
The work was published on April 30 in eLife.
The life-sustaining system for these organ linings is a supportive structure called the extracellular matrix that lies underneath the single-cell sheets. To maintain the architecture of an organ, cells that step out of line and detach from this matrix are programmed to die. "But cancer cells have gotten around this," said Elledge.
To find out how, Natasha Pavlova, a graduate student in the Elledge lab, went on a hunt for genes that allow normal cells to proliferate even when the matrix is removed. Testing a collection of 8,000 human genes, she found that the gene called PVRL4 is a strong enabler of this growth phenomenon characteristic of cancer.
PVRL4 is normally active only in a small number of tissues, such as the skin and cornea, but is frequently overabundant in breast, lung and ovarian cancers. Other research has shown that PVRL4 is found in 100 percent of breast cancers that have spread to more than one lymph node. Its presence also predicts poor outcomes for cancer patients.
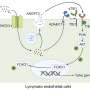
By silencing the gene with a technique called RNA interference in cells taken from several different breast tumors, Elledge and colleagues confirmed that cancer cells use the gene's activity to their advantage, staying alive even when they lose connection with the matrix. PVRL4 produces a cell-surface protein that allows the cells to stick to other cells. These cell-to-cell connections send a signal that a cell is still attached to the matrix, even when it is not. "It's as if it gives the cells a way to pack up a virtual matrix and take it with them," said Elledge.
To bind to other cells, PVRL4 seeks out a receptor, PVRL1. Unlike PVRL4, PVRL1 is found in a wide variety of cell types. This makes it possible for a cancer cell driven by PVRL4 to bind not only to another cancer cell, but to many different cell types that have PVRL1 on them. This property can potentially enable cancer cells to spread throughout the body, attaching themselves to remote tissues and forming metastases, said Elledge, who is still investigating this possibility.
Because PVRL4 creates a protein that sits on the surface of the cell and reaches out to bind to other cells, it can also be disrupted using monoclonal antibodies, proteins designed to seek out PVRL4 and disrupt its activity. Together with Christian Pallasch from Michael Hemann's laboratory at the Koch Institute for Integrative Cancer Research at MIT, the group tested to see whether injecting anti-PVRL4 antibodies into mice implanted with PVRL4-positive breast cancer cells would interfere with the tumor growth.
"We were excited to see that the antibody not only stopped tumors from growing, but also the mice did not show acute side effects from the treatment, even in skin, where PVRL4 is normally active," said Pavlova. After treatment ended, tumor growth resumed, but progressed more slowly than in untreated mice.
If the approach proves effective in humans, it could become part of the arsenal used to treat breast cancer. "We view this antibody treatment as a potential part of combination therapies," said Elledge.
Elledge is now working to understand what activates PVRL4 in cancer cells in the first place. In addition, the group has found that other well-known oncogenes, such as RAS and PI3K, also make cells stick to neighboring cells. However, in these cells, PVRL4 is not the culprit. "There must be other genes being turned on," he said. "We're trying to find out what they are and just how general this phenomenon is."
If multiple oncogenes drive cell-to-cell sticking that is advantageous for cancer cells, as it seems to be with PVRL4, Elledge and colleagues may have found a new paradigm for understanding the growth and spread of tumors in a large proportion of cancers.
More information: elife.elifesciences.org/content/2/e00358