Mechanism shown to reverse disease in arteries

A certain immune reaction is the key, not to slowing atherosclerosis like cholesterol-lowering drugs do, but instead to reversing a disease that gradually blocks arteries to cause heart attacks and strokes.
This is the finding of a study in mice led by researchers at NYU Langone Medical Center and published online June 26 in the Journal of Clinical Investigation.
The study focuses on reversing the effects of "bad cholesterol," which is deposited into the walls lining blood vessels in levels influenced by both genetics and a person's diet. By the fourth decade of life, and thanks to the chronic reaction to cholesterol, most people have inflamed "wounds" in their arteries called plaques, which when severe enough can rupture to cause blood clots that block arteries.
"Even the latest, most potent cholesterol-lowering drugs, PCSK9 inhibitors, let alone widely used statins, cannot fully reverse damage done to arteries over time, and so they can't prevent roughly 500,000 heart attacks per year in the United States," says lead study author Edward Fisher, MD, PhD, director of the Marc and Ruti Bell Vascular Biology and Disease Program at NYU Langone.
"We need the next generation of drugs to go beyond cholesterol lowering to address the immune reaction to accumulated cholesterol, and to dismantle plaques as part of reversing or regressing mature disease," says Fisher, the Leon H. Charney Professor of Cardiovascular Medicine at NYU Langone.
In years of painstaking research, the NYU Langone-led research team has zeroed in on the molecular events that occur in arteries when regression of atherosclerotic plaques is underway.
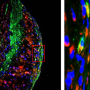
Once deposited into arteries, bad cholesterol - known to physicians as low density lipoprotein - triggers the body's immune system, which is meant to destroy invading microbes but can drive inflammatory disease in the wrong context. Immune cells in the bloodstream called monocytes swarm to cholesterol deposits, and become either inflammatory or healing cell types based on signals there.
In situations where disease is worsening in a plaque, past studies have shown that monocytes become M1 macrophages that amplify immune responses, increase inflammation, and secrete enzymes that gnaw at plaques until they rupture. The current study confirmed that monocytes arriving in plaques where disease is regressing instead become M2 "healing" macrophages, which dampen inflammation and prevent the ruptures that precede clotting.
When mice were engineered to lose the ability of monocytes to become M2 macrophages, they could no longer achieve normal disease regression, say the authors.
By surgically transplanting plaques from diseased mice into the arteries of healthy mice, the research team brought about dramatic drops in cholesterol levels. This drop has been shown to trigger a second benefit in mice, where monocytes automatically become M2 instead of M1 macrophages as plaques rapidly regress.
It is not known whether cholesterol lowering alone triggers this M2 switch in humans. But new imaging techniques may soon be able to detect changes in the type and number of macrophages in plaques. In the meantime, if researchers learn how to boost the M2 switch, a number of clinical applications may become possible just as methods arrive that can measure their success.
"A race is underway to develop treatments that enhance the decision of human monocytes to become M2 macrophages in cases where the disease has not yet caused clot formation, at which point it becomes irreversible," says Fisher.
Specifically, the current study found that the same blood-borne Ly6Chigh monocytes, once thought of only as precursors to "inflammation-prone" M1 macrophages, instead become anti-inflammatory M2 cells when they arrive in a regressing plaque. Having found the class of cells from which M2 macrophages arise, the team is now seeking to identify the local signals that tell monocytes to become M2.
Among the candidates are the immune signaling proteins interleukin-4 and interleukin-13, which have been linked by past studies to the M2 decision. These interleukins are known to turn on the STAT6 pathway, which sends this protein to the nucleus where it turns on genes that direct a monocyte to become a M2 macrophage. Researchers confirmed that blocking the action of STAT6 reduced the number of M2 macrophages in regressing plaques.
Fisher's team is already experimenting with nanoparticles based on the structure of "good cholesterol," which is known to take cholesterol of out of plaques and deliver it to the liver for destruction. One version of their nanoparticle delivers interleukin 4 to plaques as well. A next step for the work would be a study of nanoparticles in pigs, a model where success can set the stage for human trials.