Immune system 'double agent' could be new ally in cancer fight

St. Jude Children's Research Hospital scientists have discovered that an enzyme called TAK1 functions like a "double agent" in the innate immune response, serving as an unexpected regulator of inflammation and cell death. The findings highlight TAK1 inhibition as a potential cancer treatment.
TAK1 is a kinase known to promote inflammation. But Thirumala-Devi Kanneganti, Ph.D., a member of the St. Jude Children's Research Hospital Department of Immunology, and her colleagues showed that TAK1 can suppress inflammation as well. The research appeared online earlier this month in the Journal of Experimental Medicine.
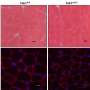
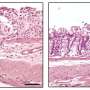
Working in mouse immune cells called macrophages, the researchers demonstrated that TAK1 plays a pivotal role in restricting inflammation and cell death by blocking spontaneous activation of the NLRP3 inflammasome. The NLRP3 inflammasome is a protein complex that, when activated, helps the innate immune system recognize and mount a rapid defense against bacteria, viruses and other threats. But elevated activation of NLRP3 is also associated with human disease, including myeloproliferative and inflammatory disorders.
NLRP3 has been the focus of more than 3,500 scientific publications since Kanneganti discovered its function more than 10 years ago. "Despite ongoing research and the central role of NLRP3 in inflammation, infection and immunity, the precise regulatory mechanism governing activation of NLRP3 and its related cell-death pathways was not clearly understood," said Kanneganti, the study's corresponding author. "These findings show that TAK1 is a central regulator of NLRP3 inflammasome homeostasis.
"The findings add to evidence that inhibitors of TAK1 and other molecules in the pathway may have a role in cancer therapy by promoting tumor cell death," she said.
NLRP3 activation normally requires two signals. TAK1's role in the process came to light when co-first author Prajwal Gurung, Ph.D., then a postdoctoral fellow in Kanneganti's laboratory and now at the University of Iowa Carver College of Medicine, made a surprising observation.
Without TAK1, macrophages derived from mouse bone marrow died spontaneously in the laboratory even in the absence of the signals normally required to activate NLRP3. Macrophages with TAK1 did not. But the normal (wild type) macrophages did die when treated with a TAK1 inhibitor.
"Such spontaneous cell death was unexpected," added first author R.K. Subbarao Malireddi, Ph.D., a scientist in Kanneganti's laboratory. "We went on to show that macrophages lacking TAK1 died due to spontaneous activation of signaling pathways that promoted NLRP3 inflammasome activation and cell death."
The consequences of TAK1 deficiency included a biochemical cascade that activated the enzymes RIPK1 and caspase-1. Caspase-1 prompts production of proteins like IL-18 and IL-1β that promote inflammation and cell death via the inflammatory cell-death pathway (pyroptosis). Researchers also detailed other molecules that drove the process in the absence of TAK1, including tumor necrosis factor.
NLRP3 is one of the five major inflammasomes involved in the innate immune response. However, Kanneganti and colleagues reported that only the NLRP3 inflammasome is spontaneously activated in macrophages in the absence of TAK1.
This study also adds to the evidence that TAK1 inhibitors may have a role in cancer immune therapy. Compounds that inhibit TAK1 stopped cell proliferation in mice, underscoring their potential in cancer treatment. Kanneganti said that is not surprising since their findings showed that, along with pyroptosis, TAK1 also inhibits apoptotic and necroptotic cell-death pathways.
"This research connects TAK1, the NLRP3 inflammasome and RIPK1, apparently for the first time, advancing our understanding of a number of key pathways," Kanneganti said.
More information: R.K. Subbarao Malireddi et al. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation, The Journal of Experimental Medicine (2018). DOI: 10.1084/jem.20171922