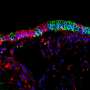
Researchers discover new type of lung cell, critical insights for cystic fibrosis
 extend through neighboring epithelial cells in the upper respiratory tract of the mouse, to the surface of the epithelial lining. Cell nuclei in cyan. Credit: Montoro et al./Nature 2018")
Researchers have identified a rare cell type in airway tissue, previously uncharacterized in the scientific literature, that appears to play a key role in the biology of cystic fibrosis. Using new technologies that enable scientists to study gene expression in thousands of individual cells, the team comprehensively analyzed the airway in mice and validated the results in human tissue.
Led by researchers from the Broad Institute of MIT and Harvard and Massachusetts General Hospital (MGH), the molecular survey also characterized gene expression patterns for other new cell subtypes. The work expands scientific and clinical understanding of lung biology, with broad implications for all diseases of the airway—including asthma, chronic obstructive pulmonary disease, and bronchitis.
Jayaraj Rajagopal, a physician in the Pulmonary and Critical Care Unit at MGH, associate member at the Broad Institute, and a Howard Hughes Medical Institute (HHMI) faculty scholar, and Broad core institute member Aviv Regev, director of the Klarman Cell Observatory at the Broad Institute, professor of biology at MIT, and an HHMI investigator, supervised the research. Daniel Montoro, a graduate student in Rajagopal's lab, and postdoctoral fellows Adam Haber and Moshe Biton in the Regev lab are co-first authors on the paper published today in Nature.
"We have the framework now for a new cellular narrative of lung disease," said Rajagopal, who is also a professor at Harvard Medical School and a principal faculty member at the Harvard Stem Cell Institute. "We've uncovered a whole distribution of cell types that seem to be functionally relevant. What's more, genes associated with complex lung diseases can now be linked to specific cells that we've characterized. The data are starting to change the way we think about lung diseases like cystic fibrosis and asthma."
"With single-cell sequencing technology, and dedicated efforts to map cell types in different tissues, we're making new discoveries—new cells that we didn't know existed, cell subtypes that are rare or haven't been noticed before, even in systems that have been studied for decades," said Regev, who is also co-chair of the international Human Cell Atlas consortium. "And for some of these, understanding and characterizing them sheds new light immediately on what's happening inside the tissue."
Using single-cell RNA sequencing, the researchers analyzed tens of thousands of cells from the mouse airway, mapping the physical locations of cell types and creating a cellular "atlas" of the tissue. They also developed a new method called pulse-seq to monitor development of cell types from their progenitors in the mouse airway. The findings were validated in human tissue.
One extremely rare cell type, making up less than one percent of the cell population in mice and humans, appeared radically different from other known cells in the dataset. The team dubbed this cell the "pulmonary ionocyte" because its gene expression pattern was similar to ionocytes—specialized cells that regulate ion transport and hydration in fish gills and frog skin.
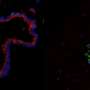
 dot the landscape of ciliated cells (magenta) of the mouse lung airway lining. Credit: Montoro et al./Nature 2018")
Strikingly, at levels higher than any other cell type, these ionocytes expressed the gene CFTR—which, when mutated, causes cystic fibrosis in humans. CFTR is critical for airway function, and for decades researchers and clinicians have assumed that it is frequently expressed at low levels in ciliated cells, a common cell type spread throughout the entire airway.
But according to the new data, the majority of CFTR expression occurs in only a few cells, which researchers didn't even know existed until now.
When the researchers disrupted a critical molecular process in pulmonary ionocytes in mice, they observed the onset of key features associated with cystic fibrosis—most notably, the formation of dense mucus. This finding underscores how important these cells are to airway-surface regulation.
"Cystic fibrosis is an amazingly well-studied disease, and we're still discovering completely new biology that may alter the way we approach it," said Rajagopal. "At first, we couldn't believe that the majority of CFTR expression was located in these rare cells, but the graduate students and postdocs on this project really brought us along with their data."
The results may also have implications for developing targeted cystic fibrosis therapies, according to the team. For example, a gene therapy that corrects for a mutation in CFTR would need to be delivered to the right cells, and a cell atlas of the tissue could provide a reference map to guide that process.
The study further highlighted where other disease-associated genes are expressed in the airway. For example, asthma development has been previously linked with a gene that encodes a sensor for rhinoviruses, and the data now indicate that this gene is expressed by ciliated cells. Another gene linked with asthma is expressed in tuft cells, which separated into at least two groups—one that senses chemicals in the airway and one that produces inflammation. The results suggest that a whole ensemble of cells may be responsible for different aspects of asthma.
Using the pulse-seq assay, the researchers tracked how the newly characterized cells and subtypes in the mouse airway develop. They demonstrated that mature cells in the airway arise from a common progenitor: the basal cells. The team also discovered a previously undescribed cellular structure in the tissue. These structures, which the researchers called "hillocks," are unique zones of rapid cell turnover, and their function is not yet understood.
"The atlas that we've created is already starting to drastically re-shape our understanding of airway and lung biology," said Regev. "And, for this and other organ systems being studied at the single-cell level, we'll have to drape everything we know on top of this new cellular diversity to understand human health and disease."
More information: Daniel T. Montoro et al, A revised airway epithelial hierarchy includes CFTR-expressing ionocytes, Nature (2018). DOI: 10.1038/s41586-018-0393-7
Related: Lindsey W. Plasschaert et al. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte, Nature (2018). DOI: 10.1038/s41586-018-0394-6