Excessive degradation of mitochondria is the tipping point from normal alcohol metabolism to alcoholic liver disease
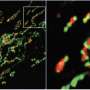
 are damaged and degraded (shown in green) upon ethanol exposure (right) that is not seen under normal conditions (left). Credit: Drs. Lemasters and Zhong / MUSC")
While most commonly known as "the powerhouses of the cell" because of their energy producing capabilities, mitochondria also play important roles in regulating the health of cells. These important structures can be damaged by alcohol consumption, which can cause them to rupture and release their DNA, proteins and lipids, collectively known as "damage associated molecular patterns (DAMPs)."
Rupture of mitochondria and release of DAMPs causes widespread inflammation, which can lead to cellular damage in the liver, contributing to liver disease and even cancer. To prevent this, cells have a process called mitophagy to remove damaged mitochondria selectively before they cause inflammation. However, the processes that regulate mitophagy and its long-term effects on liver health in conditions of alcohol exposure are less well known.
To understand more fully how alcohol damages mitochondria, and how this leads to mitophagy, researchers at the Medical University of South Carolina (MUSC) used an advanced imaging technique to investigate changes in mitochondrial function within the livers of mice that were exposed to alcohol. Their findings, published online on March 16 in the journal Autophagy, demonstrated that exposure to alcohol causes a specific type of mitochondrial damage called depolarization. In a completely novel discovery, they found that it is this depolarization that indicates to the cell that the mitochondria are damaged and thereby causes activation of the mitophagy machinery to remove the damaged mitochondria before they can cause harm.
"Mitochondria can become a timebomb, and we have to contain them," said John J. Lemasters, M.D., Ph.D., director for the Center for Cell Death, Injury and Regeneration and the GlaxoSmithKline Distinguished Endowed Chair in the College of Pharmacy.
Previously, researchers noted that after acute (or short-term) alcohol exposure—such as that which occurs during a single night of binge drinking—mitochondria are damaged but can recover. During alcohol processing in the liver, the toxic molecule acetaldehyde (AcAld) is produced, which can harm cells. Mitochondria aid in the removal of AcAld, but they become damaged or depolarized in the process, necessitating their removal. Undamaged mitochondria are replicated, and the cells continue to function properly.
Thus, as summarized by Lemasters, who originally coined the term mitophagy, damage in the form of depolarization is beneficial because it helps to rapidly remove ethanol, specifically the toxic byproduct acetaldehyde.
However, in chronic (or long-term) conditions of alcohol exposure—such as that which occurs after years of alcohol consumption as seen in alcohol addiction—the continual cycle of mitochondrial damage followed by their removal becomes pathogenic, as fewer healthy mitochondria remain each time. Consistent removal of so many injured mitochondria leads to problems such as mitochondrial depletion syndrome, where not enough ATP (or energy) is produced. Other cellular functions also begin to decline.
The current study determined that mitochondrial injury, specifically depolarization, initiates mitophagy to prevent damaged mitochondria from accumulating in cells. Blocking depolarization after ethanol exposure also blocks mitophagy, preventing mitochondrial depletion.
"No mitochondria whatsoever are better than bad mitochondria," said Lemasters.
These so-called "bad" mitochondria, which have been damaged by the toxic AcAld, can release their toxic DAMPs into the cells if not segregated and degraded. Zhi Zhong, M.D., Ph.D., professor in the department of Drug Discovery and Biomedical Sciences and senior author for this study, mentioned that DAMPs may be useful in future studies.
"Mitochondrial DAMPs are associated with liver cell damage and may be able to be used as a biomarker to see a correlation between the severity of disease and liver damage," Zhong said.
Directly inhibiting depolarization is likely to be difficult. Therefore, the team investigated methods to assist the mitochondria in processing AcAld so that fewer mitochondria are injured through depolarization and degraded. One method is to boost the activity of an enzyme that processes AcAld using a drug called Alda-1. While treatment with this drug successfully decreased the number of injured mitochondria in the livers of mice, Alda-1 has shown significant widespread toxicity in clinical trials and is therefore a poor candidate for clinical use. However, the success of Alda-1 in reducing mitochondrial injury, which may be beneficial in the long term, provides the groundwork for developing better drugs and methods for preventing mitochondrial injury in the liver.
Interestingly, the questions posed in this study could only be answered using animal models. This is because in cell culture models, mitochondrial depolarization following acute alcohol treatment does not occur. Thus, the researchers used a relatively new technology called intravital multiphoton microscopy, which is a fancy way to obtain detailed images of cellular structures in living animals. Combined with innovative mouse models that make fluorescently colored cell components, which allow their health and interaction to be tracked, previously unanswerable questions regarding the consequences of alcohol exposure on mitochondria were investigated.
The MUSC team was able to determine that both acute and chronic alcohol exposure leads to mitochondrial damage in the form of depolarization. The mitochondrial depolarization directly initiates mitophagy to degrade the damaged mitochondria. While acute injury can be repaired by replication of healthy and undamaged mitochondria, chronic exposure to alcohol causes repeated degradation of the mitochondria, leading to mitochondrial depletion, inflammation, fibrosis, cirrhosis, and ultimately, liver disease.
Determining how alcohol damages mitochondria may lead to novel methods for diagnosing early-stage liver disease and developing more effective treatments. As well as providing more detailed basic science knowledge about mitochondrial function and triggers of mitophagy, this manuscript lays the groundwork for future studies on developing novel therapies for alcoholic and nonalcoholic liver diseases.
More information: Devadoss J. Samuvel et al, Mitochondrial depolarization after acute ethanol treatment drives mitophagy in living mice, Autophagy (2022). DOI: 10.1080/15548627.2022.2046457