February 20, 2015 report
Cutting off a cancer cell at its transcriptional source: Model system for designing a small molecule inhibitor

(Medical Xpress)—What if you could attack cancer cells at their source without hurting the surrounding healthy cells? A group of researchers from the University of Virginia, the University of Massachusetts, Cornell University, and the University of Kansas constructed a small molecule inhibitor that targets a mutated protein present in leukemia cells, halting the progression of leukemia in both mouse models and in human cells. Their work appears in Science.
Acute myeloid leukemia (AML) is the most common type of leukemia in adults. Strategies to combat AML typically involve nonselective chemotherapy, which can have good short-term results but poor long-term survival. Additionally, chemotherapy can be burdensome on certain groups, including the elderly. An ideal therapy would involve targeting the mechanism producing AML cells while leaving the healthy blood cells intact. To do this, it is important to pinpoint what mechanism is causing normal blood cells to malfunction, producing leukemia cells.
There are several sub-categories of AML based on the particular genetic mutations that occur in blood cells. One of these subcategories is inv(16) AML. Inv(16)(p13q22) is an inversion factor driving the mutation that produces preleukemic progenitor cells, and following additional mutations, those cells eventually become leukemia cells. Healthy bone marrow cells, the cells that produce blood cells, have protein subunit called core-binding factor-beta (CBFB). Its job is to regulate the genes used in the production of blood and bone cells. Specifically, it binds to a RUNX1 gene, which is the transcription factor for producing blood cells.
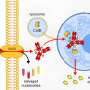
In inv(16) AML cells, CBFB is stopped from doing its normal job of regulating RUNX1. Instead CBFB is fused with smooth muscle myosin heavy chain molecule (SMMHC). The result wreaks havoc on the blood stem cell, causing it to produce unhealthy white blood cells and too few red blood cells. The CBFB-SMMHC complex is so devastating because even in the presence of normal CBFB, the complex will preferentially bind to RUNX1, deactivating the gene.
Anuradha Illendula and John A. Pulikkan et al. sought to design a small molecule that would preferentially bind to the CBFB-SMMHC complex, but would leave healthy the wild-type protein, CBFB alone, thus protecting the healthy cells. They successfully found such a molecule using a systematic strategy that could, potentially, be used in other disease models.
Their strategy involved first identifying a small molecule that would inhibit CBFB-SMMHC from binding to RUNX1 by investigating the National Cancer Institute, NIH, Diversity Set using a high-throughput fluorescence resonance energy transfer assay with Venus-CBFB-SMMHC replacing Venus-CBFB. Once they identified a good candidate, they needed to modify it. To ensure specificity to the CBFB-SMMHC complex while leaving cells with unbound CBFB alone, they decided to exploit the fact that CBFB is monomeric while CBFB-SMMHC is oligomeric by designing a bivalent molecule using varying lengths of polyethylene glycols to link two of the candidate molecules. After further testing confirmed selectivity for CBFB-SMMHC complex, they then incorporated a trifluoromethoxy (CF3O) substituent to molecule to decrease the divalent molecule's reactivity in vivo.
Illendula and Pulikkan's team eventually landed on AI-10-49 as their target molecule. In vitro studies with human AML (ME-1) cell lines showed that this molecule caused a 90% dissociation of RUNX1 from CBFB-SMMCH after six hours of treatment and had negligible activity in normal bone marrow cells, confirming selectivity. In vivo studies showed that the mice that received ten days of treatment with AI-10-49 lived, on average, eight weeks while those that did not receive treatment lived for an average of four weeks. The authors also note that although they did not conduct a formal toxicity study, they did not observe any evidence of toxicity in the treated mice.
This strategy is a proof of concept that could serve as a model for producing polyvalent small molecules that would selectively suppress ocogenic fusion proteins. Their method, while specific to homodimers, could be applied to other cancers where transcriptional dysregulation causes cancer cell proliferation.
More information: "A small molecule inhibitor of the aberrant transcription factor CBF-beta-SMMHC delays leukemia in mice" Science vol 347, issue 6223, page 779. www.sciencemag.org/content/347/6223/779
ABSTRACT
Acute myeloid leukemia (AML) is the most common form of adult leukemia. The transcription factor fusion CBFβ-SMMHC (core binding factor β and the smooth-muscle myosin heavy chain), expressed in AML with the chromosome inversion inv(16)(p13q22), outcompetes wild-type CBFβ for binding to the transcription factor RUNX1, deregulates RUNX1 activity in hematopoiesis, and induces AML. Current inv(16) AML treatment with nonselective cytotoxic chemotherapy results in a good initial response but limited long-term survival. Here, we report the development of a protein-protein interaction inhibitor, AI-10-49, that selectively binds to CBFβ-SMMHC and disrupts its binding to RUNX1. AI-10-49 restores RUNX1 transcriptional activity, displays favorable pharmacokinetics, and delays leukemia progression in mice. Treatment of primary inv(16) AML patient blasts with AI-10-49 triggers selective cell death. These data suggest that direct inhibition of the oncogenic CBFβ-SMMHC fusion protein may be an effective therapeutic approach for inv(16) AML, and they provide support for transcription factor targeted therapy in other cancers.
© 2015 Medical Xpress