Heart attack treatment hypothesis 'busted'

Researchers have long had reason to hope that blocking the flow of calcium into the mitochondria of heart and brain cells could be one way to prevent damage caused by heart attacks and strokes. But in a study of mice engineered to lack a key calcium channel in their heart cells, Johns Hopkins scientists appear to have cast a shadow of doubt on that theory. A report on their study is published online this week in Proceedings of the National Academy of Sciences.
"We confirmed that this calcium channel is important for heart function," says senior investigator Mark Anderson, M.D., Ph.D., director of the Department of Medicine at the Johns Hopkins University School of Medicine. "But our results also showed that this is almost certainly not going to be a good pathway to exploit in a long-term therapy, at least for heart attacks."
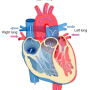
The experiments by Anderson and his team grew out of increased understanding in recent years of the role of calcium in heart function. With each beat of the organ, molecules of calcium whiz in and out of tiny compartments called mitochondria that are powerhouses of heart and other cells. Inside the mitochondria, calcium is generally a good thing—it helps generate energy that the cells use to stay alive. But for almost half a century, researchers have also known that too much mitochondrial calcium can overwhelm and cause cells to die. And after a heart attack or stroke, a sudden rush of calcium into the organelles sets off this cell death pathway, leading to long-term damage.
Thus, the possibility of saving heart and brain cells by blocking this influx of calcium, Anderson says, has long been a hope, one fed a few years ago when scientists discovered the specific channel that allows calcium to pass in and out of the mitochondria, known as the mitochondrial calcium uniporter (MCU).
With the new knowledge, Anderson and colleagues set out to test the effects of blocking calcium from mitochondria by generating genetically altered mice with a mutation that disabled heart MCU function over the entire lifetime of the mice and blocked calcium flow to mitochondria in heart muscle cells.
Although almost no calcium passed into the mitochondria of their cardiomyocytes, Anderson says, their hearts still beat and developed normally. But when his team stressed the mice in a way that would normally cause an increase in heart rate, the mice's heart rates only barely rose, and their heart muscles lost efficiency, requiring extra oxygen to function.
In further experiments, when the scientists cut off oxygen to the cardiomyocytes and then restarted it—mimicking what happens during some heart attacks—the cells still died, even though calcium in the mitochondria clearly wasn't causing the cell death, Anderson says.
Instead, the cardiomyocytes, Anderson's group discovered, were compensating for the lack of calcium by activating other cell death pathways and turning on a host of new genes to get that job done. Blocking calcium from the mitochondria, it turned out, just changed the way the cells died after a heart attack.
"Despite the predictions that blocking this calcium channel would protect against calcium overload, it didn't protect against cell death," says Anderson. Future studies will be needed to confirm whether the same is true in brain cells, but Anderson suspects the new results will put a damper on the idea of creating drugs to block MCUs in humans requiring long-term treatments to prevent heart attacks.
More information: Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart www.pnas.org/cgi/doi/10.1073/pnas.1504705112