Researchers identify new target regulating mitochondria during stress

Like an emergency response team that is called into action to save lives, stress response proteins in the heart are activated during a heart attack to help prevent cell death. As part of this process, Lewis Katz School of Medicine at Temple University researchers show for the first time that one of these specialized emergency responder proteins, known as MCUB, temporarily decreases harmful levels of calcium transport into mitochondria, the energy-generating batteries of cells.
The new research, published online September 19 in the journal Circulation, identifies MCUB as a promising new target for the investigation and treatment of conditions that feature calcium overload and cell death—conditions that include heart failure, heart attack, stroke, and neurodegeneration.
"MCUB fine-tunes calcium uptake by mitochondria in injured heart tissue, in an attempt to limit calcium overload, which is a major contributor to cell death, particularly following a heart attack," explained John W. Elrod, Ph.D., Associate Professor in the Center for Translational Medicine at the Temple University Lewis Katz School of Medicine and senior investigator on the new study.
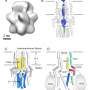
Calcium homeostasis is vital to a number of day-to-day cellular activities and is regulated primarily by mitochondria. For calcium to enter mitochondria, it passes through a channel known as the mitochondrial calcium uniporter (MCU), which resides in the inner mitochondrial membrane where it stimulates the production of ATP, the energy currency of the cell. The amount of calcium that mitochondria take up is regulated by various components of this channel. While MCUB closely resembles the pore forming subunit, MCU, its precise role in calcium regulation is largely unknown, particularly in the context of disease.
Dr. Elrod's team found that deletion of the MCUB gene in cells results in a change in the proteins that make up the calcium channel and that are essential for controlling whether the channel is on or off. Since these alterations are induced by stress, such as heart cell injury, the researchers next investigated the role of MCUB after heart attack in mice. In mice that suffered heart attack, the research team observed significant elevations in MCUB gene expression and decreases in MCU and the gatekeeper of the channel, MICU1. When genetically expressed prior to inducing a heart attack in mice, MCUB altered the channel to reduce calcium overload in the injured heart, ultimately curtailing tissue injury.
Dr. Elrod's team also found that, while it can improve cell survival after heart injury, increased MCUB activity comes at the expense of mitochondrial energy production. "MCUB induction is a compensatory change," explained Dr. Elrod. Just like an emergency responder, MCUB moves in and tries to reduce cell death and aid cell survival—however, the reduction in mitochondrial calcium uptake is also maladaptive and limits the cell's ability to increase energy during stress.
"MCUB presents us with a new molecular target for investigation," Dr. Elrod said. "It's unique in that it alters the stoichiometry of the channel and thereby presents a new mechanism which may be amendable to therapeutic manipulation. We think that modulating MCUB may allow us to tune down mitochondrial calcium uptake without completely inhibiting all energetic function."
It is hoped that follow-up studies defining the exact sites of molecular interaction will provide additional insight into how to target mitochondrial calcium overload in heart disease.