This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
In the genetics of congenital heart disease, noncoding DNA fills in some blanks
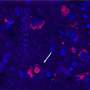
 and cardiac enhancers (Enh 5–19). Credit: Nature Genetics (2024). DOI: 10.1038/s41588-024-01669-y")
Researchers have been chipping away at the genetic causes of congenital heart disease (CHD) for a couple of decades. About 45% of cases of CHD have an identifiable cause, including chromosomal abnormalities, genetic variants affecting protein-coding genes, and environmental factors. What about the rest of the cases of CHD?
Noncoding DNA elements have long been thought to play a role in CHD, but few variants have been pinned down as causative. New work from the lab of cardiologist William Pu, MD, at Boston Children's Hospital provides the strongest evidence to date that noncoding DNA contributes to CHD. It also establishes a pipeline for finding and validating variants.
"It's very hard to figure out which noncoding variants are actually involved in heart disease and which are just innocent bystanders," says Pu, associate chair of Basic and Translational Cardiovascular Research at Boston Children's. "The average person has about 70 variants in noncoding regions that are de novo (not shared with their parents). And if you compare people with and without heart disease, they have the same number of variants."
As described in their article published in Nature Genetics, Pu and his colleagues drew on data from the multi-center Pediatric Cardiac Genomics Consortium (PCGC), led at Boston Children's by Jane Newburger, MD, MPH; Amy Roberts, MD; and Wendy Chung, MD, Ph.D.
The consortium has enrolled more than 13,000 patients with CHD (roughly a fifth of them patients at Boston Children's) and more than 18,000 family members. More than 3,000 patients have had their complete genomes sequenced, and Pu's lab obtained whole genome sequencing data for 750 patients and their parents.
Zeroing in on causative variants
To eliminate noncoding variants that were likely to be background "noise" rather than causative, the research team applied statistical filters. They also focused on de novo variants, which are more likely to be causative.
This left them with nearly 7,000 variants in noncoding DNA regions in the patients with CHD. To explore the variants' function, the team created a high-throughput system to test their effect on the expression of adjacent genes.
"Of the 7,000 variants we tested, we found 403 that affected the activity of transcription enhancers," says Pu. "These would be lead candidates for variants that may actually contribute to congenital heart disease."
They then performed a painstaking task, introducing 10 of the noncoding variants into normal human stem cells at the appropriate location in the genome. When they directed the stem cells to form cardiomyocytes, four of the 10 variants altered the expression of neighboring genes.
"Those genes were already known to cause congenital heart disease when mutated," Pu says. "The variants either made the enhancer not work as well as it should, so that the gene was expressed at a lower level, or turned a region that's not supposed to be an enhancer into an enhancer, activating a gene at the wrong time or the wrong place. Either scenario could potentially cause heart disease."
As a second test, they performed single-cell sequencing of the cardiomyocytes derived from the stem cells with the four variants. These cells also had an altered gene expression profile, consistent with the potential to cause CHD.
Future directions in CHD genetics
Pu believes that hundreds more noncoding variants may be involved in structural heart disease, helping to explain many cases of CHD that are negative by exome sequencing. Co-first author Sarah Morton, MD, Ph.D., of the Division of Newborn Medicine specializes in studying genetic causes of CHD. Using data from the study, she created an algorithm to prioritize those variants most likely to be related to CHD for further investigation.
"When we assess coding genetic variants, such as missense variants that change a single amino acid, we want to know the likelihood that they will alter protein expression," Morton says. "Relatively few tools exist to predict the functional impact of noncoding variants, particularly as they may relate to pediatric disease."
The team hopes to extend its search beyond cardiomyocytes to other cell types, such as neural crest cells and endocardial cells.
In the future, the findings could potentially be used to stratify patients' risks, predict outcomes of surgery—or even, in a more distant future, change the trajectory of structural heart disease during pregnancy or after birth.
"That is a whole unexplored area," Pu says. "We've only found the tip of the iceberg in this study."
More information: Feng Xiao et al, Functional dissection of human cardiac enhancers and noncoding de novo variants in congenital heart disease, Nature Genetics (2024). DOI: 10.1038/s41588-024-01669-y